The Mechanism of Oxygen Transfer from an Oxaziridine to a Sulfide and a Sulfoxide: a Theoretical Study
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Highly Efficient Camphor-Derived Oxaziridines for the Asymmetric
Highly Efficient Camphor-Derived Oxaziridines for the Asymmetric Oxidation of Sulfides to Chiral Sulfoxides Vassilios Meladinis, Uwe Verfürth, and Rudolf Herrmann* Organisch-Chemisches Institut der Technischen Universität München, Lichtenbergstraße 4, D-8046 Garching, Bundesrepublik Deutschland Dedicated to Prof. Dr. Ivar Ugi on the occasion o f his 60th birthday Z. Naturforsch. 45b, 1689- 1694 (1990); received May 18, 1990 Asymmetric Oxidation, N-Sulfonyl-oxaziridines, Chiral Sulfoxides, Camphorsulfonic Acid Chiral N-sulfonyl-oxaziridines derived from 8 -camphorsulfonic acid and fenchone have been evaluated as asymmetric oxidizing agents for the conversion of sulfides to chiral sulf oxides. There is no correlation between the redox potentials nor the lvO NMR chemical shifts of the oxaziridines and their relative oxidation rates, nor with the enantiomeric excesses achieved, indicating that steric effects are responsible for their behaviour. The results are con sistent with an attack of one sulfur lone pair at the oxaziridine oxygen in such a way that both sulfur lone pairs lie in the plane of the oxaziridine ring. The most efficient oxaziridines, the camphorlactone-sulfonyloxaziridine [(4aS,9aR)-10,10-dimethyl-6,7-dihydro-4H-4a,7-meth- ano-oxazirino[3,2-j]oxepino[3,4-c]isothiazol-9(5 H)-one 3,3-dioxide] and the 3-endo-bromo- camphorsulfonyloxaziridine [(4aS,8 S ,8 aR)-8-bromo-9,9-dimethyl-5,6,7,8-tetrahydro-4 H- 4a,7-methano-oxazirino-2,l-benzisothiazole 3,3-dioxide] allow the preparation of chiral sul foxides with up to 85% enantiomeric excess. Introduction crowded oxaziridines activated by an electron- Chiral sulfoxides play a prominent role among withdrawing sulfonyl group at nitrogen give the the chiral auxiliaries used for the synthesis of enan- best results. -

Synthesis of New Camphor-Based Auxiliaries
UNIVERSITY OF HAWAllllB~ PART 1: SYNTHESIS OF NEW CAMPHOR-BASED AUXILIARIES PART 2: ISOMERIZATION / CYCLIZATION OF ACETYLENIC KETONES TO CYCLOPENTENONES A THESIS SUBMITTED TO THE GRADUATE DIVISION OF THE UNIVERSITY OF HAWAI'I IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF SCIENCE IN CHEMISTRY MAY 2003 By Jeremy S. Forest Thesis Committee: Marcus A. Tius, Chairperson Thomas K. Hemscheidt Craig M. Jensen ACKNOWLEDGEMENTS I would first like to give my sincere thanks to my advisor, Dr. Marcus A. Tius. His endless guidance and support inside the laboratory are lessons that I will carry along forever in my journey through life. I would also like to thank the members of my dissertation committee for their time and effort. I would like to extend a special thanks to Dr. Thomas Hemscheidt for his tireless efforts in the review of this thesis. Many thanks go to Wesley Yoshida and Mike Burger for their help in obtaining NMR and mass spectra. I would also like to thank the members of the Tius group, especially Brad Tokeshi, Cisco Bee, Frank Cordaro, and Eric LeClerc, for their endless help and companionship during my time here. Once again, I would like to thank Dr. Marcus A. Tius for his generous financial support in the form of a research assistantship. I cannot take full credit for this work without recognizing my parents, Bill and Felicia. Their unconditional love and support has kept me going in everything that I do. Finally, I have to thank the fellas: Dave, Scott, Nick, Mitch, and brother Josh. They always believed in me and encouraged me to work through the good and the bad. -

Progress Toward the Total Synthesis of Terpenoid Natural Products: the Neomangicols and the Yohimbine Alkaloids
Progress Toward the Total Synthesis of Terpenoid Natural Products: the Neomangicols and the Yohimbine Alkaloids By Jessica Louise Wood A dissertation in submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy in Chemistry in the Graduate Division of the University of California, Berkeley Committee in charge: Professor Richmond Sarpong, Chair Professor F. Dean Toste Professor Leonard Bjeldanes Fall 2011 Abstract Progress Toward the Total Synthesis of Terpenoid Natural Products: the Neomangicols and the Yohimbine Alkaloids by Jessica Louise Wood Doctor of Philosophy in Chemistry University of California, Berkeley Professor Richmond Sarpong, Chair Progress has been made toward the total synthesis of a diverse array of natural products. Chapter 1 begins by introducing the isolation, bioactivity, and biosynthesis of the neomangicol and mangicol sesterterpenoids. Subsequent to that introduction, a summary of previous synthetic approaches to these natural products is presented. In the third section, our synthetic approaches are detailed, beginning with a first generation synthesis of the ABD tricycle, followed by a description of our revised route to the neomangicol tetracyclic core and our work toward the rearrangement of that core to the mangicol spirocyclic core. This chapter concludes with a summary of our accomplishments in this natural product area and outlines several strategies to achieve the desired rearrangement. The last section also includes our initial studies into the formation of the mangicol core. Preliminary work toward the synthesis of the ABD tricycle was performed by Dr. Brian Pujanauski. Chapter 2 details our work in the area of the yohimbine alkaloids. It begins with an introduction to these pentacyclic indole-containing natural products, discussing their isolation, proposed biosynthesis and giving a brief overview of the rich bioactivity that has been ascertained for these molecules. -

A Publication of Reliable Methods for the Preparation of Organic Compounds
A Publication of Reliable Methods for the Preparation of Organic Compounds Working with Hazardous Chemicals The procedures in Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices. In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red “Caution Notes” within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices. The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein. -

Dioxiranes: Synthesis and Reactions of Methyldioxiranes
J. Org. Chem. 1985,50, 2847-2853 2847 30 min at -15 "C, about 60% of the SO2 was removed. The liquid that at 5.54 to collapse to a 4.5 Hz d. Anal. Calcd for was stirred into diethyl ether, the ether was decanted, and acetone C14H17N3011S2:C, 35.97; H, 3.67; N, 8.99. Found: C, 36.24; H, was added to yield upon filtration 29 g (85%) of 15. 3.54; N, 8.96. Preparation of 15 in CH2C12. To 70 g of CH2C12and 20 g 1-(2-Thienyl)tetrahydrothiopheniumPicrate (17). To 75 (0.23 mol) of THT at -30 "C was added 14 g (0.40 mol) of chlorine g of SO2 and 16 g (0.18 mol) of THT at -30 "C was added 21.7 followed by addition of 18.1 g (0.17 mol) of styrene. After 30 min, g (0.16 mol) of S02C12followed by 13.96 g (0.16 mol) of thiophene. the solution was stirred into ether, the ether was decanted, and After 30 min at -5 "C 30 mL of H20 was added and the SO2 was acetone was added to give 4.2 g (9.2%) of 15: 60-MHz 'H NMR removed. After extraction twice with both chloroform and hex- (D20)6 7.5 (5 H, b, phenyl), 5.48 [l H, t, J = 7 Hz, C(2) HI, 3.96 anol, the material was converted to the picrate by the usual [2 H, AB of ABX, J(AB) = 13 Hz, C(1) H2], 3.4 (4 H, m, width manner to give 9.3 g (14.6%) of 17: 100-MHz 'H NMR 20 Hz, THT+ a H), 2.14 (4 H, m, width 14 Hz, THT' /3 H); (Me2SO-d6)6 8.61 (2 H, s, picrate), 8.22 [lH, dd, J = 5.1 Hz, J' 60-MHz 'H NMR (CF,COOH) 6 7.5 (5 H, b, phenyl), 5.45 (1 H, = 1.4 Hz, C(5) HI, 8.00, [l H, dd, J = 3.8 Hz, J'= 1.4 Hz, C(3) t, J = 7 Hz), 3.85 (2 H, t, J = 7 Hz), 3.55 (4 H, m, width 25 Hz), HI, 7.34 [l H, dd, J = 5.1 Hz, J'= 3.8 Hz, C(4) HI, 3.88 (4 H, 2.36 (4 H, m, width 15 Hz). -

Bioorganic Studies in AIDS: Synthetic Antifungals Against Pneumocystis Carinii Based on the Multivalency Concept
Int. J. Mol. Sci. 2002, 3, 1145-1161 International Journal of Molecular Sciences ISSN 1422-0067 © 2002 by MDPI www.mdpi.org/ijms/ Bioorganic Studies in AIDS: Synthetic Antifungals Against Pneumocystis carinii Based on the Multivalency Concept Langu Peng, Cunxiang Chen, Christian R. Gonzalez and Valeria Balogh-Nair* Department of Chemistry, City College of CUNY, 138 Street & Convent Avenue, New York, NY10031 Tel.: 212 650 8340, Fax: 212 650-6057, E-mail: [email protected] *Author to whom correspondence should be addressed. Received: 7 June 2002 / Accepted: 30 October 2002 / Published: 30 November 2002 Abstract: We report the syntheses of antifungals containing the novel pharmacophores: oxaziridines, sulfonyloxaziridines, nitrones and nitronyl nitroxides. We hypothesized that multiple copies of the pharmacophore per molecule might be a prerequisite to enhance efficacy against the opportunistic pathogen, Pneumocystis carinii. Therefore structural optimization of the leads was based on this new “multivalency” approach. All bisoxaziridines were inactive, but a trisoxaziridine caused ca. 50% reduction of the number of P. carinii tropozoites, compared to TMP-SMX, and a hexaoxaziridine at 1 µg/ml showed activity comparable to the currently used drug, TMP-SMX. Insertion of three units of the nitronyl nitroxide pharmacophore per molecule afforded an antifungal triradical with activity comparable to TMP-SMX at 1 µg/ml; at 25 µg/ml and at 10 µg/ml the triradical was better. The results lend further support to the oxidoredox pharmacophore hypothesis, and the enhancement of activities observed demonstrates the high potential and benefits of applying the concept of multivalency to drug development. Keywords: AIDS, opportunistic infections, Pneumocystis carinii, multivalency in drug design, antifungals, oxaziridines, sulfonyloxaziridines, nitrones, nitronyl nitroxides. -

View PDF Version
Green Chemistry View Article Online COMMUNICATION View Journal | View Issue Hydrogen peroxide/dimethyl carbonate: a green system for epoxidation of N-alkylimines and Cite this: Green Chem., 2016, 18, 4859 N-sulfonylimines. One-pot synthesis of N N Received 20th May 2016, -alkyloxaziridines from -alkylamines and Accepted 22nd July 2016 (hetero)aromatic aldehydes† DOI: 10.1039/c6gc01394e a,b b a www.rsc.org/greenchem Jamil Kraïem, Donia Ghedira and Thierry Ollevier* A green method for epoxidation of imines using an environ- and oxidation with buffered oxone.24 Perfluoro-oxaziridines mentally benign oxidant system, H2O2/dimethyl carbonate, was have also been prepared by the oxidation of the corresponding N 25 developed. -Alkyloxaziridines were prepared in high yields from perfluoro-imines with H2O2 in the presence of a base. These N 26 11,17 Creative Commons Attribution 3.0 Unported Licence. -alkylamines and (hetero)aromatic aldehydes in one-pot fashion, methods usually use toxic solvents such as CH3CN and 19b whereas N-sulfonyloxaziridines have been prepared by using the CH2Cl2. Furthermore, most of them generate stoichiometric same oxidant system and 5 mol% of Zn(OAc)2·2H2O as catalyst. amounts of undesirable waste, and require tedious work-up and purification. 1 Oxaziridines, first reported by Emmons in 1957, exhibit Recently, such synthetic routes utilizing hazardous solvents unusual and distinctive reactivity, which is linked to the and reagents and generating toxic waste have become discour- nature of their N-substituent. For example, N-sulfonyloxaziri- aged and there have been much efforts to develop safer and dines have been used as electrophilic oxygenating agents for environmentally-benign alternatives. -
![A Thesis Entitled "RING CLEAVAGE of OXAZIRID]NES" Submitted By](https://docslib.b-cdn.net/cover/4577/a-thesis-entitled-ring-cleavage-of-oxazirid-nes-submitted-by-3184577.webp)
A Thesis Entitled "RING CLEAVAGE of OXAZIRID]NES" Submitted By
A thesis entitled "RING CLEAVAGE OF OXAZIRID]NES" submitted by ANA MARIA FELIX TRINDADE LOBO in partial fulfilment of the requirements for the Degree of Doctor of Philosophy in the Faculty of Science University of London Imperial College, October,1971 London, S.W.7. i 2 ABSTRACT A general survey of the chemistry of oxaziridines is presented, previous studies on the mechanism of the acid and base catalysed hydrolysis are critically reviewed and the results discussed in relation with the hydrolysis of compounds with related structures. Results of the acid hydrolysis of several oxaziridines are reported and agree with previous findings. These accord with a mechanism involving the breakdown of an 0-conjugate acid intermediate to give either a carbonium ion or an immonium ion structure, which subsequently reacts-with water. The importance of each pathway depends on the nature of the 3-substituent. The proton of to the ring nitrogen seems to be involved in the rate determining step for the reaction of 2-primary-alkyl 3-alkyl oxaziridines. For 2-secondary-alkyl 3- alkyl compounds extensive migration of an 45( group occurs and steric requirements and migratory aptitudes seem to be important. Inves- tigations of the site of protonation suggest N as the most likely site, although hydrolysis probably proceeds via the 0-conjugate acid: pK values for oxaziridines reflect the nature of the 3-substituent A as well as the degree of branching at position 2 and lie in the range -1.9 to +0.30. Results of basic hydrolysis of several oxaziridines are reported and are consistent with earlier findings. -

Electronic Structure and Reactivity of Dioxirane and Carbonyl Oxide
J. Am. Chem. SOC.1992, 114, 7207-7217 7207 Footnotes for Table I1 (continued) l-x,-y,-z (distance 3); 0,: I/,+X,~,~/~-Zand 0,: 3/2-x,-y,z+l/2 (distance 4); OZ4: x,l+y,l+z and 026:x,l+y,z (distance 5); 020:x,l+y,l+z and OI8: I/2-x,1/2+y,1/2+z(distance 6). CDistanceacross the 12-ring aperture between those oxygen atoms that in the initial, symmetric geometry of space group P6/mmm (No. 191) are generated from 0,at 0.0000,0.2754,0.5000 and O2at 0.1659,0.3318,0.5000 by the following operations-0,: x-y,-y,z and 0,: y,y-x,z (distance 1); 02:x,y,z and 02:x-y,-y,r (distance 2). /Distance across the 12-ring aperture between those oxygen atoms that in the initial, symmetric geometry of space group P6/mmm (No. 191) are generated from 0, at 0.0000, 0.2815, 0.5000 and O2 at 0.1639, 0.3278, 0.5000 by the following operations-0,: x,y,r and 01:y,y-x,z (distance 1); 02:x,y,z and 0,: x-y,-y,z (distance 2). gDistance across the 12-ring aperture between those oxygen atoms that in the initial, symmetric geometry of space group P4,22 (No. 91) are generated from 0,1at 0.8354, 0.8390, 0.0607 and OM at 0.8356, 0.4949, 0.0608, Osoat 1.000, 0.8215, 0.000 and 060at 1.000, 0.5126, 0,0000 by the following operations-O,,: x,~-JJ,~/~-zand 040:x,y,z (distance 1); Om: I-JJ,x,'/~+zand Oso: Z-Y,X,'/~+Z(distance 2); OM: 2-x,y,l-z and OJI: 2-x.1- Y,~/~+z(distance 3); Oso:y,x,)14-z and Om: l+y,X,'/4+2 (distance 4); 03,:Z-~,X,~/~+Z and OM: l-y,~,'/~+z(distance 7); 031:x,Z-y,l/,-z and OM: ~,l-y,l/~-z(distance 8). -

New Green Technologies for Organocatalytic Asymmetric Epoxidation Applications in Synthesis
NEW GREEN TECHNOLOGIES FOR ORGANOCATALYTIC ASYMMETRIC EPOXIDATION APPLICATIONS IN SYNTHESIS Noor Armylisas Binti Abu Hassan Submitted in partial fulfillment for the requirements for the award of Doctor of Philosophy University of East Anglia School of Chemistry University of East Anglia March 2014 ©This copy of the thesis has been supplied on condition that anyone who consults it is understood to recognise that its copyright rests with the author and that no quotation from the thesis, nor any information derived there from, may be published without the author’s prior, without consent DECLARATION This thesis is submitted to the University of East Anglia for the Degree of Doctor of Philosophy and has not been previously submitted at this or any university for assessment or for any other degree. Except where stated, and reference and acknowledgement is given, this work is original and has been carried out by the author alone. NOOR ARMYLISAS BINTI ABU HASSAN ii ABSTRACT New Green Technologies for Organocatalytic Asymmetric Epoxidation Applications in Synthesis Noor Armylisas Binti Abu Hassan This thesis describes the application of asymmetric epoxidation reactions on chromene substrates mediated by Page’s iminium salt catalysts. Organocatalytic asymmetric epoxidation is an efficient tool to access enantiomerically rich epoxides. This study is divided into three main parts which are discussed in each chapter. The first part of this research is the preparation of iminium salt catalysts followed by synthesis of several chromene substrates. The final part is the application of the asymmetric epoxidation of the readily prepared iminium salts on chromene substrates. The first chapter reviews brief introduction and historical background of organocatalysis, chromene substrates and asymmetric epoxidation reactions. -
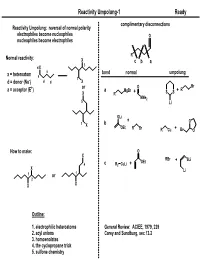
Reactivity Umpolung-1
Reactivity Umpolung-1 Ready complimentary disconnections Reactivity Umpolung: reversal of normal polarity electrophiles become nucleophiles O nucleophiles become electrophiles R Normal reactivity: X c ba 3 d X a bond normal umpolung x = heteroatom a 1 d = donor (Nu-) d d X + or O Br a = acceptor (E ) a MgBr + R X R + SS NMe2 5 Li OLi + O 1 b X OEt R Br R Cu + Br O How to make: O X RBr OEt + SLi 4 c R2 CuLi + X Li 1 or 1 2 X X Outline: 1. electrophilic heteroatoms General Review: ACIEE, 1979, 239 2. acyl anions Carey and Sundburg, sec 13.2 3. homoenolates 4. the cyclopropane trick 5. sulfone chemistry Reactivity Umpolung-2 Ready Electrophilic Heteroatoms mCPBA 'Rubottom [O]' OTMS 1. mCPBA OTMS O General form: 2. HF Ph Ph Ph 60% O OH X X Y+ X,Y = heteroatoms Y Rubottom, TL, 1974, 4319 Electrophile Examples O O O O2 O O 1. LDA; O2 O 1. TBSOTf, Et3N DMDO PO PO OTBS NMe2 2. Na2S2O3 HO 2. DMDO NMe2 84% P'O OP" 69% P'O OP" OP" O OP" HO O NMe2 Falck, JOC, 1995, 3385 Oxaziridines 1. LiHMDS 2. " HO Ph CO2Et Ph CO2Et 81% O O N OH O S O Wasserman and Lipshutz O TL, 1975, 1731 O 95% ee O O Mo O O Davis, Chem. Rev. 1992, 919 O O OH Py O LDA; MoOPH not a general asymmetric method. For racemic, most 87% commonly used is 'Davis Oxaziridine' P(NMe2)3 O MoOPH Ph Vedejs, JOC, 1978, 188 TsN Reactivity Umpolung-3 Ready Aminations (Review: Syn Lett, 1997, 741) Heteroatom exchange: Most commonly with Br, Cl Electrophile Examples O O cat PCl3 O NH3 R R R OH E OH OH Br2 OH N 1. -

Information to Users
INFORMATION TO USERS This manuscript has been reproduced from the microfilm master UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter 6 ce, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely afreet reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand comer and continuing from left to right in equal sections with small overlaps. Each original is also photographed in one exposure and is included in reduced form at the back of the book. Photographs included in the original manuscript have been reproduced xerographically in this copy. Ifigher quality 6” x 9” black and white photographic prints are available for any photographs or illustrations appearing in this copy for an additional charge. Contact UMI directly to order. UMI A Bell & Howell Infoimation Compaiqr 300 North Zedb Road, Ann Aibor Ml 48106-1346 USA 313/761-4700 800/521-0600 NOTE TO USERS The original manuscript received by UMI contains slanted print. All efforts were made to acquire the highest quality manuscript from the author or school.