View • Inclusion in Pubmed and All Major Indexing Services • Maximum Visibility for Your Research
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

A Mutation in the Putative MLH3 Endonuclease Domain Confers a Defect in Both Mismatch Repair and Meiosis in Saccharomyces Cerevisiae
Copyright Ó 2008 by the Genetics Society of America DOI: 10.1534/genetics.108.086645 A Mutation in the Putative MLH3 Endonuclease Domain Confers a Defect in Both Mismatch Repair and Meiosis in Saccharomyces cerevisiae K. T. Nishant, Aaron J. Plys and Eric Alani1 Department of Molecular Biology and Genetics, Cornell University, Ithaca, New York 14853-2703 Manuscript received January 2, 2008 Accepted for publication March 20, 2008 ABSTRACT Interference-dependent crossing over in yeast and mammalian meioses involves the mismatch repair protein homologs MSH4-MSH5 and MLH1-MLH3. The MLH3 protein contains a highly conserved metal- binding motif DQHA(X)2E(X)4E that is found in a subset of MLH proteins predicted to have endonuclease activities (Kadyrov et al. 2006). Mutations within this motif in human PMS2 and Saccharomyces cerevisiae PMS1 disrupted the endonuclease and mismatch repair activities of MLH1-PMS2 and MLH1-PMS1, re- spectively (Kadyrov et al. 2006, 2007; Erdeniz et al. 2007). As a first step in determining whether such an activity is required during meiosis, we made mutations in the MLH3 putative endonuclease domain motif (-D523N, -E529K) and found that single and double mutations conferred mlh3-null-like defects with respect to meiotic spore viability and crossing over. Yeast two-hybrid and chromatography analyses showed that the interaction between MLH1 and mlh3-D523N was maintained, suggesting that the mlh3-D523N mutation did not disrupt the stability of MLH3. The mlh3-D523N mutant also displayed a mutator phenotype in vegetative growth that was similar to mlh3D. Overexpression of this allele conferred a dominant-negative phenotype with respect to mismatch repair. -

E2013080118.Full.Pdf
Replication-independent instability of Friedreich’s ataxia GAA repeats during chronological aging Alexander J. Neila,1, Julia A. Hiseya,1, Ishtiaque Quasema, Ryan J. McGintya, Marcin Hitczenkob, Alexandra N. Khristicha, and Sergei M. Mirkina,2 aDepartment of Biology, Tufts University, Medford, MA 02155; and bFederal Reserve Bank of Atlanta, Atlanta, GA 30309 Edited by Sue Jinks-Robertson, Duke University School of Medicine, Durham, NC, and approved December 28, 2020 (received for review June 25, 2020) Nearly 50 hereditary diseases result from the inheritance of abnor- For example, in the case of Huntington’s disease (HD), an in-frame mally long repetitive DNA microsatellites. While it was originally (CAG)n repeat causes the buildup of disruptive polyglutamine believed that the size of inherited repeats is the key factor in protein aggregates in the brain with age (7, 8). In FRDA, where the disease development, it has become clear that somatic instability of causative repetitive element is noncoding, it has been hypothesized these repeats throughout an individual’s lifetime strongly contrib- that frataxin deficiency drives disease progression in a similar utes to disease onset and progression. Importantly, somatic insta- fashion via accumulation of toxic iron due to the dysregulation of bility is commonly observed in terminally differentiated, postmitotic iron homeostasis (9). An emerging alternative to the toxicity hy- cells, such as neurons. To unravel the mechanisms of repeat insta- pothesis is that the primary driver of symptom onset and progres- bility in nondividing cells, we created an experimental system to sion could be age-related, somatic expansions of the inherited ’ analyze the mutability of Friedreich s ataxia (GAA)n repeats during disease-size microsatellite. -
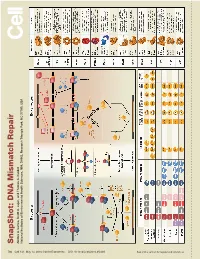
S Na P S H O T: D N a Mism a Tc H R E P a Ir
SnapShot: Repair DNA Mismatch Scott A. Lujan, and Thomas Kunkel A. Larrea, Andres Park, NC 27709, USA Triangle Health Sciences, NIH, DHHS, Research National Institutes of Environmental 730 Cell 141, May 14, 2010 ©2010 Elsevier Inc. DOI 10.1016/j.cell.2010.05.002 See online version for legend and references. SnapShot: DNA Mismatch Repair Andres A. Larrea, Scott A. Lujan, and Thomas A. Kunkel National Institutes of Environmental Health Sciences, NIH, DHHS, Research Triangle Park, NC 27709, USA Mismatch Repair in Bacteria and Eukaryotes Mismatch repair in the bacterium Escherichia coli is initiated when a homodimer of MutS binds as an asymmetric clamp to DNA containing a variety of base-base and insertion-deletion mismatches. The MutL homodimer then couples MutS recognition to the signal that distinguishes between the template and nascent DNA strands. In E. coli, the lack of adenine methylation, catalyzed by the DNA adenine methyltransferase (Dam) in newly synthesized GATC sequences, allows E. coli MutH to cleave the nascent strand. The resulting nick is used for mismatch removal involving the UvrD helicase, single-strand DNA-binding protein (SSB), and excision by single-stranded DNA exonucleases from either direction, depending upon the polarity of the nick relative to the mismatch. DNA polymerase III correctly resynthesizes DNA and ligase completes repair. In bacteria lacking Dam/MutH, as in eukaryotes, the signal for strand discrimination is uncertain but may be the DNA ends associated with replication forks. In these bacteria, MutL harbors a nick-dependent endonuclease that creates a nick that can be used for mismatch excision. Eukaryotic mismatch repair is similar, although it involves several dif- ferent MutS and MutL homologs: MutSα (MSH2/MSH6) recognizes single base-base mismatches and 1–2 base insertion/deletions; MutSβ (MSH2/MSH3) recognizes insertion/ deletion mismatches containing two or more extra bases. -

Coding and Noncoding Variants in HFM1, MLH3, MSH4, MSH5, RNF212, and RNF212B Affect Recombination Rate in Cattle
Downloaded from genome.cshlp.org on September 29, 2021 - Published by Cold Spring Harbor Laboratory Press Research Coding and noncoding variants in HFM1, MLH3, MSH4, MSH5, RNF212, and RNF212B affect recombination rate in cattle Naveen Kumar Kadri,1 Chad Harland,1,2 Pierre Faux,1 Nadine Cambisano,1,3 Latifa Karim,1,3 Wouter Coppieters,1,3 Sébastien Fritz,4,5 Erik Mullaart,6 Denis Baurain,7 Didier Boichard,5 Richard Spelman,2 Carole Charlier,1 Michel Georges,1 and Tom Druet1 1Unit of Animal Genomics, GIGA-R & Faculty of Veterinary Medicine, University of Liège (B34), 4000 Liège, Belgium; 2Livestock Improvement Corporation, Newstead, 3240 Hamilton, New Zealand; 3Genomics Platform, GIGA, University of Liège (B34), 4000 Liège, Belgium; 4Allice, 75012 Paris, France; 5GABI, INRA, AgroParisTech, Université Paris-Saclay, 78350 Jouy-en-Josas, France; 6CRV BV, 6800 AL Arnhem, the Netherlands; 7InBioS-Eukaryotic Phylogenomics, Department of Life Sciences and PhytoSYSTEMS, University of Liège (B22), 4000 Liège, Belgium We herein study genetic recombination in three cattle populations from France, New Zealand, and the Netherlands. We identify 2,395,177 crossover (CO) events in 94,516 male gametes, and 579,996 CO events in 25,332 female gametes. The average number of COs was found to be larger in males (23.3) than in females (21.4). The heritability of global recombi- nation rate (GRR) was estimated at 0.13 in males and 0.08 in females, with a genetic correlation of 0.66 indicating that shared variants are influencing GRR in both sexes. A genome-wide association study identified seven quantitative trait loci (QTL) for GRR. -

Mammalian BLM Helicase Is Critical for Integrating Multiple Pathways of Meiotic Recombination
JCB: Article Mammalian BLM helicase is critical for integrating multiple pathways of meiotic recombination J. Kim Holloway, Meisha A. Morelli, Peter L. Borst, and Paula E. Cohen Department of Biomedical Sciences, College of Veterinary Medicine, Cornell University, Ithaca, NY 14853 loom’s syndrome (BS) is an autosomal recessive prompting us to explore the meiotic phenotype of mice disorder characterized by growth retardation, bearing a conditional mutant allele of Blm. In this study, B cancer predisposition, and sterility. BS mutated we show that BLM deficiency does not affect entry into (Blm), the gene mutated in BS patients, is one of five mam- prophase I but causes severe defects in meiotic pro- malian RecQ helicases. Although BLM has been shown gression. This is exemplified by improper pairing and to promote genome stability by assisting in the repair of synapsis of homologous chromosomes and altered pro- DNA structures that arise during homologous recombina- cessing of recombination intermediates, resulting in tion in somatic cells, less is known about its role in meiotic increased chiasmata. Our data provide the first analy- recombination primarily because of the embryonic lethal- sis of BLM function in mammalian meiosis and strongly ity associated with Blm deletion. However, the localiza- argue that BLM is involved in proper pairing, synapsis, tion of BLM protein on meiotic chromosomes together with and segregation of homologous chromosomes; how- evidence from yeast and other organisms implicates a ever, it is dispensable for the accumulation of recombi- role for BLM helicase in meiotic recombination events, nation intermediates. Introduction In humans, mutation of the RecQ helicase Bloom’s syndrome indicating that this protein may play an important role in mei (BS) mutated (BLM) results in the carrier suffering from the otic progression. -

Application of Genomic Technologies to Study Infertility Nicholas Rui Yuan Ho Washington University in St
Washington University in St. Louis Washington University Open Scholarship Arts & Sciences Electronic Theses and Dissertations Arts & Sciences Spring 5-15-2016 Application of Genomic Technologies to Study Infertility Nicholas Rui Yuan Ho Washington University in St. Louis Follow this and additional works at: https://openscholarship.wustl.edu/art_sci_etds Part of the Bioinformatics Commons, Genetics Commons, and the Molecular Biology Commons Recommended Citation Yuan Ho, Nicholas Rui, "Application of Genomic Technologies to Study Infertility" (2016). Arts & Sciences Electronic Theses and Dissertations. 786. https://openscholarship.wustl.edu/art_sci_etds/786 This Dissertation is brought to you for free and open access by the Arts & Sciences at Washington University Open Scholarship. It has been accepted for inclusion in Arts & Sciences Electronic Theses and Dissertations by an authorized administrator of Washington University Open Scholarship. For more information, please contact [email protected]. WASHINGTON UNIVERSITY IN ST. LOUIS Division of Biology and Biomedical Sciences Computational and Systems Biology Dissertation Examination Committee: Donald Conrad, Chair Barak Cohen Joseph Dougherty John Edwards Liang Ma Application of Genomic Technologies to Study Infertility by Nicholas Rui Yuan Ho A dissertation presented to the Graduate School of Arts & Sciences of Washington University in partial fulfillment of the requirements for the degree of Doctor of Philosophy May 2016 St. Louis, Missouri © 2016, Nicholas Rui Yuan Ho Table of -

Expression of the Mutl Homologue Hmlh3 in Human Cells and Its Role in DNA Mismatch Repair
Research Article Expression of the MutL Homologue hMLH3 in Human Cells and its Role in DNA Mismatch Repair Elda Cannavo,1 Giancarlo Marra,1 Jacob Sabates-Bellver,1 Mirco Menigatti,2 Steven M. Lipkin,3 Franziska Fischer,1 Petr Cejka,1 and Josef Jiricny1 1Institute of Molecular Cancer Research, University of Zurich, Zurich, Switzerland; 2Department of Internal Medicine, Faculty of Medicine, University of Modena, Modena, Italy; and 3Departments of Medicine and Biological Chemistry, University of California, Irvine, Irvine, California Abstract could be corroborated by analysis of the phenotypes of MMR- The human mismatch repair (MMR) proteins hMLH1 and deficient cells: Those lacking hMSH2 are fully MMR deficient and hPMS2 function in MMR as a heterodimer. Cells lacking either display a mutator phenotype and microsatellite instability that is protein have a strong mutator phenotype and display micro- consistent with the loss of repair of both base-base mismatches satellite instability, yet mutations in the hMLH1 gene account and insertion/deletion loops. Cells lacking hMSH6 retain a strong for f50% of hereditary nonpolyposis colon cancer families, mutator phenotype but their microsatellite instability is limited to mononucleotide repeats due to the functional redundancy with whereas hPMS2 mutations are substantially less frequent and h less penetrant. Similarly, in the mouse model, Mlh1À/À animals hMutS in insertion/deletion loop repair. This situation is mirrored are highly cancer prone and present with gastrointestinal in hereditary nonpolyposis colon cancer families, where the À/À penetrance of hMSH2 mutations is substantially higher than that tumors at an early age, whereas Pms2 mice succumb to cancer much later in life and do not present with gastro- of alterations in the hMSH6 locus (reviewed in ref. -

Content Based Search in Gene Expression Databases and a Meta-Analysis of Host Responses to Infection
Content Based Search in Gene Expression Databases and a Meta-analysis of Host Responses to Infection A Thesis Submitted to the Faculty of Drexel University by Francis X. Bell in partial fulfillment of the requirements for the degree of Doctor of Philosophy November 2015 c Copyright 2015 Francis X. Bell. All Rights Reserved. ii Acknowledgments I would like to acknowledge and thank my advisor, Dr. Ahmet Sacan. Without his advice, support, and patience I would not have been able to accomplish all that I have. I would also like to thank my committee members and the Biomed Faculty that have guided me. I would like to give a special thanks for the members of the bioinformatics lab, in particular the members of the Sacan lab: Rehman Qureshi, Daisy Heng Yang, April Chunyu Zhao, and Yiqian Zhou. Thank you for creating a pleasant and friendly environment in the lab. I give the members of my family my sincerest gratitude for all that they have done for me. I cannot begin to repay my parents for their sacrifices. I am eternally grateful for everything they have done. The support of my sisters and their encouragement gave me the strength to persevere to the end. iii Table of Contents LIST OF TABLES.......................................................................... vii LIST OF FIGURES ........................................................................ xiv ABSTRACT ................................................................................ xvii 1. A BRIEF INTRODUCTION TO GENE EXPRESSION............................. 1 1.1 Central Dogma of Molecular Biology........................................... 1 1.1.1 Basic Transfers .......................................................... 1 1.1.2 Uncommon Transfers ................................................... 3 1.2 Gene Expression ................................................................. 4 1.2.1 Estimating Gene Expression ............................................ 4 1.2.2 DNA Microarrays ...................................................... -

Exo1 Recruits Cdc5 Polo Kinase to Mutlγ to Ensure Efficient Meiotic Crossover Formation
Exo1 recruits Cdc5 polo kinase to MutLγ to ensure efficient meiotic crossover formation Aurore Sancheza,b,c, Céline Adama,b, Felix Rauhd, Yann Duroca,b, Lepakshi Ranjhac, Bérangère Lombarde, Xiaojing Muf,g, Mélody Wintreberta,b, Damarys Loewe, Alba Guarnéh, Stefano Gnana,b, Chun-Long Chena,b, Scott Keeneyf,g,i, Petr Cejkac, Raphaël Guéroisj, Franz Kleind, Jean-Baptiste Charbonnierj, and Valérie Bordea,b,1 aInstitut Curie, Paris Sciences et Lettres Research University, CNRS, UMR3244, 75005 Paris, France; bParis Sorbonne Université, UMR3244, 75005 Paris, France; cInstitute for Research in Biomedicine, Faculty of Biomedical Sciences, Università della Svizzera Italiana, 6501 Bellinzona, Switzerland; dMax F. Perutz Laboratories, University of Vienna, 1010 Wien, Austria; eInstitut Curie, Paris Sciences et Lettres Research University, 75006 Paris, France; fMolecular Biology Program, Memorial Sloan Kettering Cancer Center, New York, NY 10065; gWeill Graduate School of Medical Sciences, Cornell University, New York, NY 10065; hDepartment of Biochemistry, Centre de Recherche en Biologie Structurale, McGill University, Montreal, QC H3A 0G4, Canada; iMemorial Sloan Kettering Cancer Center, Howard Hughes Medical Institute, New York, NY 10065; and jInstitute for Integrative Biology of the Cell (I2BC), Commissariat à l’Energie Atomique, CNRS, Université Paris-Sud, Université Paris-Saclay, 91190 Gif-sur-Yvette, France Edited by Anne M. Villeneuve, Stanford University, Stanford, CA, and approved October 14, 2020 (received for review July 7, 2020) Crossovers generated during the repair of programmed meiotic recruits a MutL-related complex to initiate repair together with double-strand breaks must be tightly regulated to promote accu- proliferating cell nuclear antigen and Exo1 (reviewed in ref. 11). rate homolog segregation without deleterious outcomes, such as MutLα represents the major mismatch repair activity, which in- aneuploidy. -

Direct Association of Bloom's Syndrome Gene Product with The
4378–4386 Nucleic Acids Research, 2001, Vol. 29, No. 21 © 2001 Oxford University Press Direct association of Bloom’s syndrome gene product with the human mismatch repair protein MLH1 Graziella Pedrazzi, Claudia Perrera1, Heiko Blaser, Patrick Kuster, Giancarlo Marra1, Sally L. Davies2, Gi-Hyuck Ryu, Raimundo Freire3,IanD.Hickson2, Josef Jiricny1 and Igor Stagljar* Institute of Veterinary Biochemistry and Molecular Biology, University of Zürich, Winterthurerstrasse 190, CH-8057 Zürich, Switzerland, 1Institute of Medical Radiobiology of the University of Zürich and the Paul Scherrer Institute, August Forel-Strasse 7, CH-8008 Zürich, Switzerland, 2Imperial Cancer Research Fund Laboratiories, Institute of Molecular Medicine, University of Oxford, John Radcliffe Hospital, Oxford OX3 9DS, UK and 3Unidad de Investigacion, Hospital Universitario de Canarias, Ofra s/n, La Cuesta, 38071 Tenerife, Spain Received July 9, 2001; Revised and Accepted September 14, 2001 ABSTRACT chromosome breaks and sister chromatid exchanges (SCEs) Bloom’s syndrome (BS) is a rare genetic disorder (10). Consistent with this, mutations in the S.cerevisiae SGS1 and S.pombe rqh1+ genes produce phenotypes that resemble characterised by genomic instability and cancer those observed in cells derived from BS patients: inactivation susceptibility. BLM, the gene mutated in BS, encodes of Sgs1p results in an increase in DNA recombination and amemberoftheRecQ family of DNA helicases. Here, enhanced chromosome mis-segregation (11), while rqh1p we identify hMLH1, which is involved in mismatch deficiency correlates with hypersensitivity to DNA damage repair (MMR) and recombination, as a protein that (5). Further evidence of a role for the RecQ family of helicases directly interacts with BLM both in vivo and in vitro, in homologous recombination comes from the observations and that the two proteins co-localise to discrete that RecQ, Sgs1p, BLM and WRN all disrupt four-way junc- nuclear foci. -

Human Mutlγ, the MLH1–MLH3 Heterodimer, Is an Endonuclease That Promotes DNA Expansion
Human MutLγ, the MLH1–MLH3 heterodimer, is an endonuclease that promotes DNA expansion Lyudmila Y. Kadyrovaa, Vaibhavi Gujara, Vickers Burdettb, Paul L. Modrichb,c,1, and Farid A. Kadyrova,1 aDepartment of Biochemistry and Molecular Biology, Southern Illinois University School of Medicine, Carbondale, IL 62901; bDepartment of Biochemistry, Duke University Medical Center, Durham, NC 27710; and cHoward Hughes Medical Institute, Duke University Medical Center, Durham, NC 27710 Contributed by Paul L. Modrich, December 13, 2019 (sent for review August 23, 2019; reviewed by Thomas A. Kunkel, Sergei M. Mirkin, and Darren G. Monckton) MutL proteins are ubiquitous and play important roles in DNA carry a relatively small number of the repeat units and are rela- metabolism. MutLγ (MLH1–MLH3 heterodimer) is a poorly under- tively stable (32). In contrast, disease-associated alleles have a stood member of the eukaryotic family of MutL proteins that has larger number of the repeat units and are prone to further ex- been implicated in triplet repeat expansion, but its action in this pansion (32, 33). It is also known that the longer disease- deleterious process has remained unknown. In humans, triplet re- associated alleles cause more severe symptoms (33, 34). Repeat peat expansion is the molecular basis for ∼40 neurological disor- expansions occur in both germ line and somatic tissues (33, 35– ders. In addition to MutLγ, triplet repeat expansion involves the 37). Whereas germ line expansions are responsible for the β – mismatch recognition factor MutS (MSH2 MSH3 heterodimer). phenomenon of anticipation, somatic expansions contribute to γ We show here that human MutL is an endonuclease that nicks the tissue specificity and the progressive nature of the repeat DNA.