Copper-Catalyzed (Transfer) Hydrogenation Reactions and Coupling Reactions by Intercepting Reactive Inter- Mediates Thereof
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Binuclear Copper(I) Borohydride Complex Containing Bridging Bis
crystals Article Binuclear Copper(I) Borohydride Complex Containing Bridging Bis(diphenylphosphino) Methane Ligands: Polymorphic Structures of 2 [(µ2-dppm)2Cu2(η -BH4)2] Dichloromethane Solvate Natalia V. Belkova 1 ID , Igor E. Golub 1,2 ID , Evgenii I. Gutsul 1, Konstantin A. Lyssenko 1, Alexander S. Peregudov 1, Viktor D. Makhaev 3, Oleg A. Filippov 1 ID , Lina M. Epstein 1, Andrea Rossin 4 ID , Maurizio Peruzzini 4 and Elena S. Shubina 1,* ID 1 A. N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences (INEOS RAS), 119991 Moscow, Russia; [email protected] (N.V.B.); [email protected] (I.E.G.); [email protected] (E.I.G.); [email protected] (K.A.L.); [email protected] (A.S.P.); [email protected] (O.A.F.); [email protected] (L.M.E.) 2 Inorganic Chemistry Department, Peoples’ Friendship University of Russia (RUDN University), 117198 Moscow, Russia 3 Institute of Problems of Chemical Physics, Russian Academy of Sciences (IPCP RAS), 142432 Moscow, Russia; [email protected] 4 Istituto di Chimica dei Composti Organometallici Consiglio Nazionale delle Ricerche (ICCOM CNR), 50019 Sesto Fiorentino, Italy; [email protected] (A.R.); [email protected] (M.P.) * Correspondence: [email protected]; Tel.: +7-495-135-5085 Academic Editor: Sławomir J. Grabowski Received: 18 September 2017; Accepted: 17 October 2017; Published: 20 October 2017 Abstract: Bis(diphenylphosphino)methane copper(I) tetrahydroborate was synthesized by ligands exchange in bis(triphenylphosphine) copper(I) tetrahydroborate, and characterized by XRD, FTIR, NMR spectroscopy. According to XRD the title compound has dimeric structure, [(µ2-dppm)2Cu2(η2-BH4)2], and crystallizes as CH2Cl2 solvate in two polymorphic forms (orthorhombic, 1, and monoclinic, 2) The details of molecular geometry and the crystal-packing pattern in polymorphs were studied. -

Cuh-Catalyzed Enantioselective Alkylation of Indole Derivatives with Ligand-Controlled Regiodivergence
CuH-Catalyzed Enantioselective Alkylation of Indole Derivatives with Ligand-Controlled Regiodivergence The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Ye, Yuxuan et al. "CuH-Catalyzed Enantioselective Alkylation of Indole Derivatives with Ligand-Controlled Regiodivergence." Journal of the American Chemical Society 141, 9 (January 2019): 3901–3909 © 2019 American Chemical Society As Published http://dx.doi.org/10.1021/jacs.8b11838 Publisher American Chemical Society (ACS) Version Author's final manuscript Citable link https://hdl.handle.net/1721.1/126070 Terms of Use Article is made available in accordance with the publisher's policy and may be subject to US copyright law. Please refer to the publisher's site for terms of use. HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author J Am Chem Manuscript Author Soc. Author Manuscript Author manuscript; available in PMC 2020 March 06. Published in final edited form as: J Am Chem Soc. 2019 March 06; 141(9): 3901–3909. doi:10.1021/jacs.8b11838. CuH-Catalyzed Enantioselective Alkylation of Indole Derivatives with Ligand-Controlled Regiodivergence Yuxuan Ye1, Seoung-Tae Kim2,3, Jinhoon Jeong2,3, Mu-Hyun Baik2,3,*, and Stephen L. Buchwald1,* 1Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts, 02139, United States 2Department of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon, 34141, Republic of Korea 3Center for Catalytic Hydrocarbon Functionalizations, Institute for Basic Science (IBS), Daejeon, 34141, Republic of Korea Abstract Enantioenriched molecules bearing indole-substituted stereocenters form a class of privileged compounds in biological, medicinal, and organic chemistry. -

Copper Hydride Catalyzed Hydroamination of Alkenes and Alkynes
Copper Hydride Catalyzed Hydroamination of Alkenes and Alkynes The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Pirnot, Michael T., Yi-Ming Wang, and Stephen L. Buchwald. “Copper Hydride Catalyzed Hydroamination of Alkenes and Alkynes.” Angewandte Chemie International Edition 55.1 (2016): 48–57. As Published http://dx.doi.org/10.1002/anie.201507594 Publisher Wiley Blackwell Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/110464 Terms of Use Creative Commons Attribution-Noncommercial-Share Alike Detailed Terms http://creativecommons.org/licenses/by-nc-sa/4.0/ HHS Public Access Author manuscript Author Manuscript Author ManuscriptAngew Chem Author Manuscript Int Ed Engl Author Manuscript . Author manuscript; available in PMC 2016 March 08. Published in final edited form as: Angew Chem Int Ed Engl. 2016 January 4; 55(1): 48–57. doi:10.1002/anie.201507594. Copper Hydride-Catalyzed Hydroamination of Alkenes and Alkynes Dr. Michael T. Pirnot†, Dr. Yi-Ming Wang†, and Prof. Dr. Stephen L. Buchwald Department of Chemistry, Massachusetts Institute of Technology 77 Massachusetts Avenue, Cambridge, MA 02139 (USA) Stephen L. Buchwald: [email protected] Abstract Over the past few years, CuH-catalyzed hydroamination has been discovered and developed as a robust and conceptually novel approach for the synthesis of enantioenriched secondary and tertiary amines. The success in this area of research was made possible through the large body of precedent in copper(I) hydride catalysis and the well-explored use of hydroxylamine esters as electrophilic amine sources in related copper-catalyzed processes. -

Sop Pyrophoric 2 12/16/2019
Owner DOC. NO. REV. DATE C.H.O SOP PYROPHORIC 2 12/16/2019 DOC. TITLE SOP FOR PYROPHORIC CHEMICALS Environmental Health & Safety STANDARD OPERATING PROCEDURES (SOP) FOR WORKING WITH PYROPHORIC CHEMICALS AT AMHERST COLLEGE ___________________________________________________________________ General Information Pyrophoric Chemicals are solid, liquid, or gas compounds that, when exposed to air or moisture at or below 54°C (130°F), can spontaneously ignite. Examples of Pyrophoric chemicals used at Amherst College Laboratories include: sodium hydride, zinc powder, and Grignard reagents. See the “Appendix” page below for a full list of Pyrophoric Chemicals. Pyrophoric chemicals are often used as catalysts in chemical reactions or as reducing and deprotonating agents in organic chemistry. Note that Pyrophoric chemicals may also be characterized by other hazards, hence, users of these chemicals may also need to refer to other SOPs that cover other hazards. In addition, each individual chemical’s Safety Data Sheet (SDS) should be consulted before they are used. _____________________________________________________________________________________ Personal Protective Equipment When working with Pyrophoric Chemicals, the following personal protective equipment (PPE) must be worn, at a minimum. Depending on the specific chemical, other forms of protection might be required. Consult the SDS for each chemical before use: Splash goggles Lab coat (Fire resistant lab coat highly recommended) Long pants Close toed shoes Gloves – Nitrile gloves adequate for accidental contact with small quantities. However, the use of fire resistant Nomex/ Leather Pilot’s gloves is highly recommended _____________________________________________________________________________________ Safety Devices All work with Pyrophoric chemicals must be done in a glove box, vacuum manifold, or any enclosed inert environment. If work must be done in a fume hood, ensure that the hood sash is in the lowest feasible position. -

Metal Clusters History
METAL CLUSTER BY DR. SHEERIN MASROOR For M.Sc Sem (II) Clusters in Chemistry • These are assembly of atoms or molecules which are intermediate in size between a molecule and a bulk solid. • The can be made up of diverse in stoichiometry. (Mingos and Wales 1990, Introduction to Cluster Chemistry) • Examples: Fullerene, Diborane. Metal Cluster • These are compounds that contain metal-metal bonds. • A compound that contains a group of two or more atoms where direct and substantial metal bonding is present. (Huheey Inorganic Chemistry Huheey, JE,Harper and Row, New York) • Examples: Co2(CO)8, Rh2(OAc)4, Stryker’s reagent(Osborn Complex). Dicobalt octacarbonyl Rhodium(II) acetate hexameric copper hydride ligatedwith triphenylphosphi ne Metal Clusters History • 12th century - Historically Metal cluster was calomel (Hg2Cl2). • 1826 - First polyoxometallate discovered was ammonium 3− phosphomolybdate, containing the PMo12O 40 anion, discovered by Berzelius. • 1908 - Miolatti-Rosenheim explained about the structure of 12- molybdophosphoric acid as a polyoxometalate cluster. • 1928 - Linus Pauling suggested structure that accurately accounts for structure of polyoxotungstate anion. • 1934 - J. F. Keggin obtains first crystal structure of polyoxo- metallate. Now known as Keggin stucture. • 1953 - X-Ray Evidence for Metal-to-Metal Bonds in Cupric and Chromous Acetate. -2 • 1964 - F. A. Cotton (Wilkinson Student) discovers Re2Cl8 which was having metal-metal bond. • Early 1960s - Cotton coins terms cluster when referring to chemical entities that contain one or more metal-metal bond. Preparation of Metal Clusters • Hydrothermal Synthesis • Crystallization of substances from high temperature conditions. • Generally reactions are carried in aqueous media which run in steel pressure vessels in an autoclave. -

Using Alcohols As Simple H2-Equivalents for Copper-Catalysed Transfer Semihydrogenations of Cite This: Chem
ChemComm View Article Online COMMUNICATION View Journal | View Issue Using alcohols as simple H2-equivalents for copper-catalysed transfer semihydrogenations of Cite this: Chem. Commun., 2019, 55,13410 alkynes† Received 26th August 2019, Accepted 15th October 2019 Trinadh Kaicharla, Birte M. Zimmermann, Martin Oestreich and Johannes F. Teichert * DOI: 10.1039/c9cc06637c rsc.li/chemcomm Copper(I)/N-heterocyclic carbene complexes enable a transfer copper hydride-based catalytic transformation, namely the semi- semihydrogenation of alkynes employing simple and readily available reduction of internal alkynes to alkenes.10–12 alcohols such as isopropanol. The practical overall protocol circumvents The alkyne semihydrogenation poses challenges in stereo- theuseofcommonlyemployedhighpressureequipmentwhenusing selectivity (E vs. Z) and chemoselectivity (overreduction of the 10 Creative Commons Attribution 3.0 Unported Licence. dihydrogen (H2) on the one hand, and avoids the generation of stoichio- formed alkene to the corresponding alkane, Scheme 1), and thus metric silicon-based waste on the other hand, when hydrosilanes are serves as an ideal model reaction for catalyst development. To probe used as terminal reductants. a possible transfer of a hydrogen atom from the a-position of an alcohol by means of a copper(I) catalyst, we investigated the transfer Catalytic transfer hydrogenations (TH) are attractive for the semihydrogenation of internal alkyne 1,usinga,a-dideuterated reduction of a variety of functional groups, as they offer practical benzyl alcohol (2-d2, Scheme 1). Employing readily available [SIMes- protocols circumventing the need for high pressure equipment.1 CuCl]13 in catalytic amounts (10 mol%) and NaOtBu as base, full In general, for catalytic TH there is a need for easy-to-use and safe conversion either with microwave or conventional heating to the dihydrogen (H2) sources, as these reactants are associated with the corresponding alkene 3 was found. -

II Reduction Reactions
II Reduction Reactions Objectives By the end of this section you will: 1) be able to exploit the differences in reactivity of various reducing agents (hydride vs neutral reductants) in chemoselective reductions and be able to provide a mechanistic rationale to account for their differing reactivities; 2) be able to use the inherent chirality in a substrate to control the outcome of a reduction of proximal ketones to generate selectively syn and anti 1,3- and 1,2-diols; 3) be able to rationalise the outcome of these diastereoselective reactions using T.S. diagrams; 4) have gained an appreciation of the versatility of transition metals in reduction reactions; 5) have gained an appreciation of the synthetic utility of dissolving metal reductions; 6) be able to use radical chemistry for deoxygenation and reduction of halides. II.A Reduction of Carboxylic Acid Derivatives and Related Functionality OR' H ROH RO RO carboxylic acid aldehyde primary alcohol derivatives R N RNH2 RNO2 Issues of Reactivity and Selectivity Similar issues of selectivity and reactivity to those we encountered in the case of oxidation reactions also arise in reduction reactions. 1. Chemoselectivity. Many different functional groups can be reduced in a variety of ways. We often need to selectively reduce one functional group whilst leaving others intact (remember year 1 practical!). NaBH4 Sn, HCl OH O O O2N O2N H2N Chemoselective reductions from a practical in CHM1C3 2. In the case of carboxylic acid derivatives there are two possible reduction products: an aldehyde and an alcohol. Ideally we need methods for selectively accessing either product. -

Gold Hydride Complexes: Synthesis, Structure, and Reactivity
Gold Hydride Complexes: Synthesis, Structure, and Reactivity Geoffrey Chambers Literature Seminar December 4, 2012 The preparation of transition metal hydride complexes is well established for most of the transition elements. However, isolation of gold hydride complexes has been especially elusive and, compared to the other transition metal hydrides, are sparsely represented in the literature despite being invoked in catalytic cycles.1 Cationic gold is quite oxidizing (Au3+/1+= 1.36 V, Au3+/0 = 1.52 V, Au+/0 = 1.83 V),2 and it has long been presumed that gold hydride bonds are labile due to the apparent mismatch of an oxidizing metal and a reducing ligand. The binary gold hydrides are known but are unstable with respect to the formation of gold metal and dihydrogen. These complexes have been detected only by the means of the codeposition of gold vapor with hydrogen in an argon matrix at 5 K.3 Gold complexes are known to catalyze a vast array of reactions, but the metal center typically functions merely as a Lewis acid.4 In a few cases however, gold hydride species are proposed as intermediates.5 Prior to the syntheses of the first gold hydride complexes, there was no evidence that these species were stable enough to exist in solution. Scheme 1: Syntheses of the first monomeric gold hydride complexes The first AuI hydride complex (2) was synthesized by the treatment of the chloride complex (1) with LiHBEt3 (scheme 1). The complex was confirmed to be a monomeric Au hydride in the solid state through X-ray crystallography.6 The stability of 2 is attributed to the strong σ-donor N-hetercyclic carbene (NHC) ligand and its synthesis was inspired by the dimeric 7 copper hydride NHC complex [(IPr)CuH]2 . -

Organic Superconductors with High Transition Temperatures and High
United States Patent im [ii] 3,944,578 Wolf et al. [45] Mar. 16, 1976 [54] ORGANIC SUPERCONDUCTORS WITH 2,542,481 2/1951 Crandall et al 260/397.1 HIGH TRANSITION TEMPERATURES AND HIGH CRITICAL MAGNETIC FIELDS Primary Examiner—Elbert L. Roberts [75] Inventors: Alfred A. Wolf; Ernest H. Halpern, Attorney, Agent, or Firm—R. S. Sciascia; Q. E. Hodges; D. McGiehan both of Annapolis, Md. [73] Assignee: The United States of America as [57] ABSTRACT represented by the Secretary of the Organic compounds exhibit superconducting-like be- Navy, Washington, D.C. havior, as to magnetic and electrical properties, at ele- [22] Filed: Nov. 23, 1973 vated temperatures above 21°K, where 21°K is the transition temperature of most known metallic super- [21] Appl. No.: 418,337 conducting materials. The structure of the organic ma- terials according to this invention is a plurality of su- [52] U.S. CI 260/397.1; 252/108; 252/518; perconducting clusters, forming islands within a ma- 260/478 trix of insulating material. The ratio of the clusters to [51] Int. CI.2 C07J9/00 the matrix material is a minimum at 1:104. The or- [58] Field of Search 260/397.1 ganic compound comprises two distinct atomic groups termed an R group and COOM group combining as [56] References Cited R—COOM with the COOM group clustering to form UNITED STATES PATENTS superconducting islands, within the R material matrix. 2,429,899 10/1947 Sondern et al 260/397.1 15 Claims, 6 Drawing Figures U.5. fatent March 16, 1976 Sheet 1 of 3 3,^44,578 U.S. -

Enantioselective Cuh-Catalyzed Hydroacylation Employing Unsaturated Carboxylic Acids As Aldehyde Surrogates
Enantioselective CuH-Catalyzed Hydroacylation Employing Unsaturated Carboxylic Acids as Aldehyde Surrogates The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Zhou, Yujing et al. “Enantioselective CuH-Catalyzed Hydroacylation Employing Unsaturated Carboxylic Acids as Aldehyde Surrogates.” Journal of the American Chemical Society 139, 24 (June 2017): 8126–8129 © 2017 American Chemical Society As Published http://dx.doi.org/10.1021/JACS.7B04937 Publisher American Chemical Society (ACS) Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/117062 Terms of Use Article is made available in accordance with the publisher's policy and may be subject to US copyright law. Please refer to the publisher's site for terms of use. HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author J Am Chem Manuscript Author Soc. Author Manuscript Author manuscript; available in PMC 2017 September 06. Published in final edited form as: J Am Chem Soc. 2017 June 21; 139(24): 8126–8129. doi:10.1021/jacs.7b04937. Enantioselective CuH-Catalyzed Hydroacylation Employing Unsaturated Carboxylic Acids as Aldehyde Surrogates Yujing Zhou, Jeffrey S. Bandar, and Stephen L. Buchwald* Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139 Abstract The direct asymmetric copper hydride (CuH)-catalyzed coupling of α,β-unsaturated carboxylic acids to aryl alkenes is reported to access chiral α-aryl dialkyl ketones. A variety of substrate substitution patterns, sensitive functional groups and heterocycles are tolerated in this reaction, which significantly expands the range of accessible products compared to existing hydroacylation methodology. Although mechanistic studies are ongoing, we propose that CuH-catalyzed silylation of unsaturated acids occurs to access a uniquely effective acyl electrophilic coupling partner. -
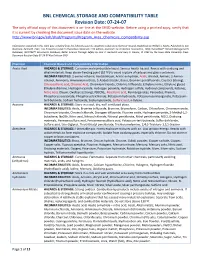
BNL CHEMICAL STORAGE and COMPATIBILITY TABLE Revision Date: 07-24-07 the Only Official Copy of This Document Is On-Line at the SHSD Website
BNL CHEMICAL STORAGE AND COMPATIBILITY TABLE Revision Date: 07-24-07 The only official copy of this document is on-line at the SHSD website. Before using a printed copy, verify that it is current by checking the document issue date on the website. http://www.bnl.gov/esh/shsd/Programs/Program_Area_Chemicals_Compatibility.asp Information contained in this table was compiled from the following sources: Academic Laboratory Chemical Hazards Guidebook by William J. Mahn, Published by Van Nostrand, Reinhold, 1991; Fire Protection Guide to Hazardous Materials 11th edition, National Fire Protection Association, 1994; Hazardtext® Hazard Managements Database; INFOTEXT® Documents Database; Better Science Through Safety by Jack A. Gerlovich and Gary E. Downs, © 1981 by the Iowa State University Press. Document Revision Date 07-24-07 Ken Erickson CHO Chemical Chemical Hazard and Compatibility Information Acetic Acid HAZARDS & STORAGE: Corrosive and combustible liquid. Serious health hazard. Reacts with oxidizing and alkali materials. Keep above freezing point (62 ºF) to avoid rupture of carboys and glass containers. INCOMPATIBILITIES: 2-amino-ethanol, Acetaldehyde, Acetic anhydride, Acids, Alcohol, Amines, 2-Amino- ethanol, Ammonia, Ammonium nitrate, 5-Azidotetrazole, Bases, Bromine pentafluoride, Caustics (strong), Chlorosulfonic acid, Chromic Acid, Chromium trioxide, Chlorine trifluoride, Ethylene imine, Ethylene glycol, Ethylene diamine, Hydrogen cyanide, Hydrogen peroxide, Hydrogen sulfide, Hydroxyl compounds, Ketones, Nitric Acid, Oleum, Oxidizers -

Anti-Markovnikov Hydroarylation of Alkenes with Aryl Halides and Synthesis And
Anti-Markovnikov Hydroarylation of Alkenes with Aryl Halides and Synthesis and Characterization of a Monomeric Copper Hydride complex Andrea Chong A thesis submitted in a partial fulfillment of the requirements for the degree of Master of Science University of Washington 2018 Committee: Dr. Gojko Lalic Dr. Michael Heinekey Program Authorized to offer Degree: Department of Chemistry © Copyright 2018 Andrea Chong Abstract Anti-Markovnikov Hydroarylation of Alkenes with Aryl Halides and Synthesis and Characterization of a Monomeric Copper Hydride complex Andrea Chong Chairperson of the Supervisory Committee: Professor Gojko Lalic Department of Chemistry Chapter 1 describes the nickel-catalyzed hydroarylation of alkenes. This reaction involves reductive cross coupling of alkenes and aryl halides in the presence of a silane as the hydride donor. High anti-Markovnikov selectivity is achieved in this reaction, and broad functional group compatibility demonstrates its utility, coupling a wide range of alkene substrates, such as alkyl- and aryl-substituted alkenes as well as enol ethers and diene. Chapter 2 describes efforts to synthesize monomeric copper hydride complexes. Preparation and characterization of various copper complexes using tris(pyrazolyl)borate (Tp), - methane (Tpm), - ethane (Tpe) and N-heterocyclic carbene (NHC) ligands were performed. Reaction of Tp ligated copper (II) methoxide complex with silane showed evidence of the existence of copper (II) hydride in the reaction. Table of Contents List of Abbreviations……………………………………………………………………………...ii