Cellular Models and in Vitro Assays for the Screening of Modulators of P-Gp, MRP1 and BCRP
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

DHEA: Dehydroepiandrosterone
DHEA: Dehydroepiandrosterone Joseph Pepping, Pharm.D. [Am J Health-Syst Pharm 57(22):2048-2056, 2000. © 2000 ASHP, Inc.] Introduction Dehydroepiandrosterone (DHEA) and its active metabolite, DHEA sulfate (DHEAS), are endogenous hormones synthesized and excreted primarily by the zona reticularis of the adrenal cortex in response to adrenocorticotropic hormone. The exact mechanism of action and clinical role, if any, of DHEA and DHEAS remain unclear. Epidemiological data indicate an inverse relationship between serum DHEA and DHEAS levels and the frequency of cancer, cardiovascular disease (in men only), Alzheimer's disease and other age-related disorders, immune function, and progression of HIV infection. [1] Animal (primarily rodent) studies have suggested many beneficial effects of DHEA, including improved immune function and memory and prevention of atherosclerosis, cancer, diabetes, and obesity. Many of the benefits seen in animal studies have yet to be shown in humans. [1-3] Uses Clinically substantiated (yet still controversial) uses of DHEA include replacement therapy in patients with low serum DHEA levels secondary to chronic disease, adrenal exhaustion, or corticosteroid therapy; treating systemic lupus erythematosus (SLE), improving bone density in postmenopausal women; improving symptoms of severe depression; improving depressed mood and fatigue in patients with HIV infection; and increasing the rate of reepithelialization in patients undergoing autologous skin grafting for burns. [1,4-8] Other possible uses (with some supporting clinical studies) include enhancing the immune response and sense of well-being in the elderly, decreasing certain cardiovascular risk factors, and treating male erectile dysfunction. [4,8-12] Use of DHEA to slow or reverse the aging process, improve cognitive function, promote weight loss, increase lean muscle mass, or slow the progression of Parkinson's disease and Alzheimer's disease is clinically unsubstantiated. -

Eurartesim, INN-Piperaquine & INN-Artenimol
ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS 1 1. NAME OF THE MEDICINAL PRODUCT Eurartesim 160 mg/20 mg film-coated tablets. 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Each film-coated tablet contains 160 mg piperaquine tetraphosphate (as the tetrahydrate; PQP) and 20 mg artenimol. For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Film-coated tablet (tablet). White oblong biconvex film-coated tablet (dimension 11.5x5.5mm / thickness 4.4mm) with a break-line and marked on one side with the letters “S” and “T”. The tablet can be divided into equal doses. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Eurartesim is indicated for the treatment of uncomplicated Plasmodium falciparum malaria in adults, adolescents, children and infants 6 months and over and weighing 5 kg or more. Consideration should be given to official guidance on the appropriate use of antimalarial medicinal products, including information on the prevalence of resistance to artenimol/piperaquine in the geographical region where the infection was acquired (see section 4.4). 4.2 Posology and method of administration Posology Eurartesim should be administered over three consecutive days for a total of three doses taken at the same time each day. 2 Dosing should be based on body weight as shown in the table below. Body weight Daily dose (mg) Tablet strength and number of tablets per dose (kg) PQP Artenimol 5 to <7 80 10 ½ x 160 mg / 20 mg tablet 7 to <13 160 20 1 x 160 mg / 20 mg tablet 13 to <24 320 40 1 x 320 mg / 40 mg tablet 24 to <36 640 80 2 x 320 mg / 40 mg tablets 36 to <75 960 120 3 x 320 mg / 40 mg tablets > 75* 1,280 160 4 x 320 mg / 40 mg tablets * see section 5.1 If a patient vomits within 30 minutes of taking Eurartesim, the whole dose should be re-administered; if a patient vomits within 30-60 minutes, half the dose should be re-administered. -

Taking Artemisinin to Clinical Anticancer Applications: Design, Synthesis and Characterization
Taking Artemisinin to Clinical Anticancer Applications: Design, Synthesis and Characterization of pH-responsive Artemisinin Dimer Derivatives in Lipid Nanoparticles Yitong Zhang A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2015 Reading Committee: Tomikazu Sasaki, Chair Rodney J.Y. Ho Champak Chatterjee Program Authorized to Offer Degree: Chemistry i ©Copyright 2015 Yitong Zhang ii University of Washington Abstract Taking Artemisinin to Clinical Anticancer Applications: Design, Synthesis and Characterization of pH-responsive Artemisinin Dimer Derivatives in Lipid Nanoparticles Yitong Zhang Chair of the Supervisory Committee: Professor Tomikazu Sasaki Chemistry iii Abstract Qinghaosu or Artemisinin is an active sesquiterpene lactone isolated from Artemisia annua L. The natural product and its derivatives are known as a first line treatment for malaria. Investigations have also reported that the compound exhibits anti-cancer activities both on cell lines and in animal models. The remarkably stable endoperoxide bridge under ambient conditions is believed to be responsible for the selectivity as well as potency against cells that are rich in iron content. Dimeric derivatives where two artemisinin units are covalently bonded through lactone carbon (C10) show superior efficacies against both malaria parasites and cancer cells. Artemisinin dimer succinate derivative demonstrates a 100-fold enhancement in potency, compared to the natural product, with IC50 values in the low micromolar range. This work focuses on the development of artemisinin dimer derivatives to facilitate their clinical development. Novel pH-responsive artemisinin dimers were synthesized to enhance the aqueous solubility of the pharmacophore motif. Compounds with promising potency against human breast cancer cell lines were selected for lipid and protein based nanoparticle formulations for delivery of the derivatives without the need of organic co-solvents into animal models. -

Factors Influencing Pesticide Resistance in Psylla Pyricola Foerster and Susceptibility Inits Mirid
AN ABSTRACT OF THE THESIS OF: Hugo E. van de Baan for the degree ofDoctor of Philosopbv in Entomology presented on September 29, 181. Title: Factors Influencing Pesticide Resistance in Psylla pyricola Foerster and Susceptibility inits Mirid Predator, Deraeocoris brevis Knight. Redacted for Privacy Abstract approved: Factors influencing pesticide susceptibility and resistance were studied in Psylla pyricola Foerster, and its mirid predator, Deraeocoris brevis Knight in the Rogue River Valley, Oregon. Factors studied were at the biochemical, life history, and population ecology levels. Studies on detoxification enzymes showed that glutathione S-transferase and cytochrome P-450 monooxygenase activities were ca. 1.6-fold higherin susceptible R. brevis than in susceptible pear psylla, however, esterase activity was ca. 5-fold lower. Esterase activity was ca. 18-fold higher in resistant pear psylla than in susceptible D. brevis, however, glutathione S-transferase and cytochrome P-450 monooxygenase activities were similar. Esterases seem to be a major factor conferring insecticideresistance in P. Pvricola. Although the detoxification capacities of P. rivricola and D. brevis were quite similar, pear psylla has developed resistance to many insecticides in the Rogue River Valley, whereas D. brevis has remained susceptible. Biochemical factors may be important in determining the potential of resistance development, however, they are less important in determining the rate at which resistance develops. Computer simulation studies showed that life history and ecological factors are probably of greater importancein determining the rate at which resistance develops in P. pvricola and D. brevis. High fecundity and low immigration of susceptible individuals into selected populations appear to be major factors contributing to rapid resistance development in pear psylla compared with D. -

Exerts Anxiolytic-Like Effects Through GABAA Receptors in a Surgical Menopause Model in Rats
Biomedicine & Pharmacotherapy 109 (2019) 2387–2395 Contents lists available at ScienceDirect Biomedicine & Pharmacotherapy journal homepage: www.elsevier.com/locate/biopha Original article Chrysin (5,7-dihydroxyflavone) exerts anxiolytic-like effects through GABAA receptors in a surgical menopause model in rats T ⁎ Juan Francisco Rodríguez-Landaa,b, , Fabiola Hernández-Lópezc, Jonathan Cueto-Escobedoa, Emma Virginia Herrera-Huertad, Eduardo Rivadeneyra-Domínguezb, Blandina Bernal-Moralesa,b, Elizabeth Romero-Avendañod a Laboratorio de Neurofarmacología, Instituto de Neuroetología, Universidad Veracruzana, Xalapa, Veracruz, Mexico b Facultad de Química Farmacéutica Biológica, Universidad Veracruzana, Xalapa, Veracruz, Mexico c Hospital General de Zona con Medicina Familiar No. 28, Delegación Veracruz Norte, Instituto Mexicano del Seguro Social (H.G.Z. c/mf. No. 28, Delegación Veracruz Norte, IMSS), Martínez de la Torre, Veracruz, Mexico d Facultad de Ciencias Químicas, Universidad Veracruzana, Orizaba, Veracruz, Mexico ARTICLE INFO ABSTRACT Keywords: The present study investigated the effects of the flavonoid chrysin (5,7-dihydroxyflavone) on anxiety-like be- Anxiolytics havior in rats in a model of surgical menopause and evaluated the participation of γ-aminobutyric acid-A Chrysin (GABAA) receptors in these actions. At 12 weeks post-ovariectomy, the effects of different doses of chrysin (0.5, GABAA 1, 2, and 4 mg/kg) were evaluated in the elevated plus maze, light/dark test, and locomotor activity test, and Oophorectomy comparisons were made with the clinically effective anxiolytic diazepam. The participation of GABA receptors Ovariectomy A in the actions of chrysin was explored by pretreating the rats with the noncompetitive GABA chloride ion Surgical menopause A channel antagonist picrotoxin (1 mg/kg). The results showed that chrysin (2 and 4 mg/kg) reduced anxiety-like behavior in both the elevated plus maze and light/dark test, and these effects were similar to diazepam. -

Clotrimazole Loaded Ufosomes for Topical Delivery: Formulation Development and In-Vitro Studies
molecules Article Clotrimazole Loaded Ufosomes for Topical Delivery: Formulation Development and In-Vitro Studies Pradeep Kumar Bolla 1 , Carlos A. Meraz 1, Victor A. Rodriguez 1, Isaac Deaguero 1, Mahima Singh 2 , Venkata Kashyap Yellepeddi 3,4 and Jwala Renukuntla 5,* 1 Department of Biomedical Engineering, College of Engineering, The University of Texas at El Paso, 500 W University Ave, El Paso, TX 79968, USA 2 Department of Pharmaceutical Sciences, University of the Sciences in Philadelphia, Philadelphia, PA 19104, USA 3 Division of Clinical Pharmacology, Department of Pediatrics, University of Utah, Salt Lake City, UT 84112, USA 4 Department of Pharmaceutics and Pharmaceutical Chemistry, College of Pharmacy, University of Utah, Salt Lake City, UT 84112, USA 5 Department of Basic Pharmaceutical Sciences, Fred Wilson School of Pharmacy, High Point University, High Point, NC 27240, USA * Correspondence: [email protected] Received: 9 August 2019; Accepted: 28 August 2019; Published: 29 August 2019 Abstract: Global incidence of superficial fungal infections caused by dermatophytes is high and affects around 40 million people. It is the fourth most common cause of infection. Clotrimazole, a broad spectrum imidazole antifungal agent is widely used to treat fungal infections. Conventional topical formulations of clotrimazole are intended to treat infections by effective penetration of drugs into the stratum corneum. However, drawbacks such as poor dermal bioavailability, poor penetration, and variable drug levels limit the efficiency. The present study aims to load clotrimazole into ufosomes and evaluate its topical bioavailability. Clotrimazole loaded ufosomes were prepared using cholesterol and sodium oleate by thin film hydration technique and evaluated for size, polydispersity index, and entrapment efficiency to obtain optimized formulation. -

TRP Mediation
molecules Review Remedia Sternutatoria over the Centuries: TRP Mediation Lujain Aloum 1 , Eman Alefishat 1,2,3 , Janah Shaya 4 and Georg A. Petroianu 1,* 1 Department of Pharmacology, College of Medicine and Health Sciences, Khalifa University of Science and Technology, Abu Dhabi 127788, United Arab Emirates; [email protected] (L.A.); Eman.alefi[email protected] (E.A.) 2 Center for Biotechnology, Khalifa University of Science and Technology, Abu Dhabi 127788, United Arab Emirates 3 Department of Biopharmaceutics and Clinical Pharmacy, Faculty of Pharmacy, The University of Jordan, Amman 11941, Jordan 4 Pre-Medicine Bridge Program, College of Medicine and Health Sciences, Khalifa University of Science and Technology, Abu Dhabi 127788, United Arab Emirates; [email protected] * Correspondence: [email protected]; Tel.: +971-50-413-4525 Abstract: Sneezing (sternutatio) is a poorly understood polysynaptic physiologic reflex phenomenon. Sneezing has exerted a strange fascination on humans throughout history, and induced sneezing was widely used by physicians for therapeutic purposes, on the assumption that sneezing eliminates noxious factors from the body, mainly from the head. The present contribution examines the various mixtures used for inducing sneezes (remedia sternutatoria) over the centuries. The majority of the constituents of the sneeze-inducing remedies are modulators of transient receptor potential (TRP) channels. The TRP channel superfamily consists of large heterogeneous groups of channels that play numerous physiological roles such as thermosensation, chemosensation, osmosensation and mechanosensation. Sneezing is associated with the activation of the wasabi receptor, (TRPA1), typical ligand is allyl isothiocyanate and the hot chili pepper receptor, (TRPV1), typical agonist is capsaicin, in the vagal sensory nerve terminals, activated by noxious stimulants. -
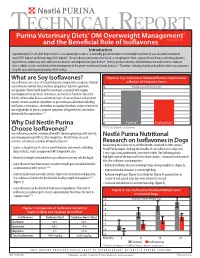
What Are Soy Isoflavones? Figure 2: Soy Isoflavones Reduced Plasma Isoprostanes Soy Isoflavones Are a Class of Natural Bioactive Compounds in Soybeans
TECHNICAL REPORT Purina Veterinary Diets® OM Overweight Management® brand canine dry formula and the Beneficial Role of Isoflavones Introduction Approximately 35% of adult dogs in the U.S. are overweight or obese1. A markedly greater incidence of overweight and obesity was observed in neutered male (59% higher) and female dogs (40% higher)1. Chronic obesity can increase the risk of, or complications from, several chronic diseases including diabetes, hypertension, pulmonary and cardiovascular disease, and degenerative joint disease2. Obesity produces chronic, mild inflammation and increases oxidative stress, which, in turn, contributes to the development of the above-mentioned chronic diseases.3,4 Therefore, reducing obesity and oxidative stress can promote a long life span and improved quality of life in dogs. What are Soy Isoflavones? Figure 2: Soy Isoflavones Reduced Plasma Isoprostanes Soy isoflavones are a class of natural bioactive compounds in soybeans. Natural a Marker of Oxidative Stress soy isoflavones include three chemical compounds: daidzein, genistein, 5 Plasma Isoprostanes (ng/ml) and glycitein. Many health benefits have been associated with regular consumption of soy products. In humans, soy has been found to reduce the 4 risk of cardiovascular disease and certain types of cancer (breast and prostate cancer); relieve a number of problems in post menopausal women including 3 hot flashes, osteoporosis, and decline in cognitive function; reduce cholesterol and triglycerides in plasma; improve symptoms of hypertension; and reduce 2 abdominal fat accumulation.5,6,7 1 Why Did Nestlé Purina 0 Control Isoflavones* Choose Isoflavones? *P<0.01 for Control vs. Isoflavones Soy isoflavones provide a number of benefits for managing dogs with obesity, and reducing rebound effects after weight loss. -

Applications of in Silico Methods to Analyze the Toxicity and Estrogen T Receptor-Mediated Properties of Plant-Derived Phytochemicals ∗ K
Food and Chemical Toxicology 125 (2019) 361–369 Contents lists available at ScienceDirect Food and Chemical Toxicology journal homepage: www.elsevier.com/locate/foodchemtox Applications of in silico methods to analyze the toxicity and estrogen T receptor-mediated properties of plant-derived phytochemicals ∗ K. Kranthi Kumara, P. Yugandharb, B. Uma Devia, T. Siva Kumara, N. Savithrammab, P. Neerajaa, a Department of Zoology, Sri Venkateswara University, Tirupati, 517502, India b Department of Botany, Sri Venkateswara University, Tirupati, 517502, India ARTICLE INFO ABSTRACT Keywords: A myriad of phytochemicals may have potential to lead toxicity and endocrine disruption effects by interfering Phytochemicals with nuclear hormone receptors. In this examination, the toxicity and estrogen receptor−binding abilities of a QSAR modeling set of 2826 phytochemicals were evaluated. The endpoints mutagenicity, carcinogenicity (both CAESAR and ISS Toxicity models), developmental toxicity, skin sensitization and estrogen receptor relative binding affinity (ER_RBA) Nuclear hormone receptor binding were studied using the VEGA QSAR modeling package. Alongside the predictions, models were providing pos- Self−Organizing maps sible information for applicability domains and most similar compounds as similarity sets from their training Clustering and classification schemes sets. This information was subjected to perform the clustering and classification of chemicals using Self−Organizing Maps. The identified clusters and their respective indicators were considered as potential hotspot structures for the specified data set analysis. Molecular screening interpretations of models wereex- hibited accurate predictions. Moreover, the indication sets were defined significant clusters and cluster in- dicators with probable prediction labels (precision). Accordingly, developed QSAR models showed good pre- dictive abilities and robustness, which observed from applicability domains, representation spaces, clustering and classification schemes. -

Estroquench™ Hormone Specific Formulation™
PRODUCT DATA DOUGLAS LABORATORIES® 08/2014 1 EstroQuench™ Hormone Specific Formulation™ DESCRIPTION EstroQuench™ is a Hormone Specific Formulation™ of ingredients that have documented anti-aromatase activity as well as androgenic adaptogens which support the function of endogenous aromatase inhibitors. Collectively these herbs promote minimal production and function of estrogens, while promoting testosterone function, including optimal sexual function in both genders. This formulation is designed to quench excessive production of estrogens and aberrant functions of them while supporting optimal function of androgens by maintaining the health of androgen producing glands.† Hormone Specific Formulation™ provided by Douglas Laboratories® and formulated by Dr. Joseph J Collins is created to support the optimal function of specific hormones through the use of hormone specific adaptogens, hormone specific agonists and hormone specific functional mimetics. This formulation may be used as part of a hormone health program with dietary and nutrient support. In addition, this formulation may be used by clinicians as an adjuvant to support optimal hormone health in patients who have been prescribed bioidentical hormone therapies. FUNCTIONS Aromatase (a cytochrome P450 enzyme {CYP19}) is the enzyme that controls the conversion of androgens to estrogens. More specifically, aromatase is the enzyme responsible for catalyzing the biosynthesis of androstenedione into estrone, and the biosynthesis of testosterone to estradiol. Estrogens include the broad range of aromatized hormones created form androgens. The specific attribute of estrogens that separate them from progestogens, androgens and corticoids is that estrogens are the only aromatized steroid hormones. Estradiol, the most potent endogenous estrogen, is biosynthesized by aromatization from androgens by aromatase (which is also called estrogen synthase). -

Natural Products As Alternative Choices for P-Glycoprotein (P-Gp) Inhibition
Review Natural Products as Alternative Choices for P-Glycoprotein (P-gp) Inhibition Saikat Dewanjee 1,*, Tarun K. Dua 1, Niloy Bhattacharjee 1, Anup Das 2, Moumita Gangopadhyay 3, Ritu Khanra 1, Swarnalata Joardar 1, Muhammad Riaz 4, Vincenzo De Feo 5,* and Muhammad Zia-Ul-Haq 6,* 1 Advanced Pharmacognosy Research Laboratory, Department of Pharmaceutical Technology, Jadavpur University, Raja S C Mullick Road, Kolkata 700032, India; [email protected] (T.K.D.); [email protected] (N.B.); [email protected] (R.K.); [email protected] (S.J.) 2 Department of Pharmaceutical Technology, ADAMAS University, Barasat, Kolkata 700126, India; [email protected] 3 Department of Bioechnology, ADAMAS University, Barasat, Kolkata 700126, India; [email protected] 4 Department of Pharmacy, Shaheed Benazir Bhutto University, Sheringal 18050, Pakistan; [email protected] 5 Department of Pharmacy, Salerno University, Fisciano 84084, Salerno, Italy 6 Environment Science Department, Lahore College for Women University, Jail Road, Lahore 54600, Pakistan * Correspondence: [email protected] (S.D.); [email protected] (V.D.F.); [email protected] (M.Z.-U.-H.) Academic Editor: Maria Emília de Sousa Received: 11 April 2017; Accepted: 15 May 2017; Published: 25 May 2017 Abstract: Multidrug resistance (MDR) is regarded as one of the bottlenecks of successful clinical treatment for numerous chemotherapeutic agents. Multiple key regulators are alleged to be responsible for MDR and making the treatment regimens ineffective. In this review, we discuss MDR in relation to P-glycoprotein (P-gp) and its down-regulation by natural bioactive molecules. P-gp, a unique ATP-dependent membrane transport protein, is one of those key regulators which are present in the lining of the colon, endothelial cells of the blood brain barrier (BBB), bile duct, adrenal gland, kidney tubules, small intestine, pancreatic ducts and in many other tissues like heart, lungs, spleen, skeletal muscles, etc. -

Test Report Comprehensive Hormone Insights™
698814 COMPREHENSIVE HORMONE INSIGHTS™ TEST REPORT Dr. Maximus, N.D. E: [email protected] Date of Collection: P: 403-241-4500 Time of Collection: F: 403-241-4501 Date of Receipt: www.rmalab.com Reported On: CHI Accession: 698814 Healthcare Professional Patient Age: Dr. Maximus, N.D. Date of Birth: Gender: Male F: Relevant Medications Biometrics Curcumin Height (in) : 73 Weight (lb) : 180 BMI : 24 Waist (in) : 35 Hip (in) : 41 CHI Accession: 698814 SUMMARY HMUS01 How to read the graphs LEGEND: 50 66 Sex Steroid Hormones 50 66 Middle third of 33 33 84 reference population Hormone Start of 83 100 80 100 highest 16 Percentile Precursors 16 Percentile third of Sum of Androgens Sum of Estrogens 50 66 reference 0 0 population (T, DHT, α+β androstanediol) Listed in Interp Guide 33 84 End of 100 16 lowest Percentile00 50 66 50 66 third of 33 33 84 reference 0 population Patient’s percentile rank 81 100 95 100 compared to reference 16 Percentile 16 Percentile population (see summary) DHEA + Metabolites Sum of Progesterone Metabolites 0 (DHEA + A + E) 0 α+β Pregnanediol Cortisol Melatonin Oxidative Stress Free Cortisol Profile (ng/mg) 100 50 66 50 66 33 84 33 84 80 64 100 0 100 16 Percentile 16 Percentile 60 6-sulfatoxy 8-Hydroxy-2- 0 Melatonin 0 deoxyguanosine 40 (Overnight) (Overnight) 20 6-sulfatoxymelatonin provides 8-hydroxy-2-deoxyguanosine is Cortisol/Creatinine (ng/mg) insight into melatonin levels. a marker of oxidative stress 0 Morning Dinner Bedtime A B C 50 66 Free cortisol Cortisol Metabolites 33 84 profile is used to provides a general Testosterone Cortisol assess diurnal assessment of 16 100 cortisol rhythm adrenal cortisol 16 Percentile Cortisol production Cortisol Metabolites 0 (α+β THF + THE) Testosterone Cortisol/Testosterone provides insight into relative catabolic (cortisol) and anabolic (testosterone) states.