The Connectome Is Necessary but Not Sufficient for the Spread of Alpha-Synuclein Pathology in Rats
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pharnygeal Arch Set - Motor USMLE, Limited Edition > Neuroscience > Neuroscience
CNs 5, 7, 9, 10 - Pharnygeal Arch Set - Motor USMLE, Limited Edition > Neuroscience > Neuroscience PHARYNGEAL ARCH SET, CNS 5, 7, 9, 10 • They are derived from the pharyngeal (aka branchial) arches • They have special motor and autonomic motor functions CRANIAL NERVES EXIT FROM THE BRAINSTEM CN 5, the trigeminal nerve exits the mid/lower pons.* CN 7, the facial nerve exits the pontomedullary junction.* CN 9, the glossopharyngeal nerve exits the lateral medulla.* CN 10, the vagus nerve exits the lateral medulla.* CRANIAL NERVE NUCLEI AT BRAINSTEM LEVELS Midbrain • The motor trigeminal nucleus of CN 5. Nerve Path: • The motor division of the trigeminal nerve passes laterally to enter cerebellopontine angle cistern. Pons • The facial nucleus of CN 7. • The superior salivatory nucleus of CN 7. Nerve Path: • CN 7 sweeps over the abducens nucleus as it exits the brainstem laterally in an internal genu, which generates a small bump in the floor of the fourth ventricle: the facial colliculus • Fibers emanate from the superior salivatory nucleus, as well. Medulla • The dorsal motor nucleus of the vagus, CN 10 • The inferior salivatory nucleus, CN 9 1 / 3 • The nucleus ambiguus, CNs 9 and 10. Nerve Paths: • CNs 9 and 10 exit the medulla laterally through the post-olivary sulcus to enter the cerebellomedullary cistern. THE TRIGEMINAL NERVE, CN 5  • The motor division of the trigeminal nerve innervates the muscles of mastication • It passes ventrolaterally through the cerebellopontine angle cistern and exits through foramen ovale as part of the mandibular division (CN 5[3]). Clinical Correlation - Trigeminal Neuropathy THE FACIAL NERVE, CN 7  • The facial nucleus innervates the muscles of facial expression • It spans from the lower pons to the pontomedullary junction. -

Probing Forebrain to Hindbrain Circuit Functions in Xenopus
Received: 15 November 2016 | Accepted: 16 November 2016 DOI 10.1002/dvg.22999 REVIEW Probing forebrain to hindbrain circuit functions in Xenopus Darcy B. Kelley1 | Taffeta M. Elliott2 | Ben J. Evans3 | Ian C. Hall4 | Elizabeth C. Leininger5 | Heather J. Rhodes6 | Ayako Yamaguchi7 | Erik Zornik8 1Department of Biological Sciences, Columbia University, New York, New York Abstract 10027 The vertebrate hindbrain includes neural circuits that govern essential functions including breath- 2Department of Psychology, New Mexico ing, blood pressure and heart rate. Hindbrain circuits also participate in generating rhythmic motor Tech, Socorro, New Mexico 87801 patterns for vocalization. In most tetrapods, sound production is powered by expiration and the 3 Department of Biology, McMaster circuitry underlying vocalization and respiration must be linked. Perception and arousal are also University, Hamilton, Ontario, Ontario linked; acoustic features of social communication sounds—for example, a baby’scry—can drive L8S4K1, Canada autonomic responses. The close links between autonomic functions that are essential for life and 4Department of Biology, Benedictine University, Lisle, Illinois vocal expression have been a major in vivo experimental challenge. Xenopus provides an opportu- 5Department of Biology, St. Mary’s College, nity to address this challenge using an ex vivo preparation: an isolated brain that generates vocal St. Mary’s City, Maryland 29686 and breathing patterns. The isolated brain allows identification and manipulation of hindbrain vocal 6Department of Biology, Denison University, circuits as well as their activation by forebrain circuits that receive sensory input, initiate motor Granville, Ohio 43023 patterns and control arousal. Advances in imaging technologies, coupled to the production of Xen- 7 Department of Biology, University of Utah, opus lines expressing genetically encoded calcium sensors, provide powerful tools for imaging Salt Lake City, Utah 84112 neuronal patterns in the entire fictively behaving brain, a goal of the BRAIN Initiative. -

Adrenergic Activation of Cardiac Preganglionic Neurons in Nucleus
bioRxiv preprint doi: https://doi.org/10.1101/276998; this version posted January 8, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Adrenergic agonist induces rhythmic firing in quiescent cardiac preganglionic neurons in nucleus ambiguous via activation of intrinsic membrane excitability Isamu Aiba and Jeffrey L. Noebels Department of Neurology, Baylor College of Medicine Houston TX 77030 Correspondence: Department of Neurology, Baylor College of Medicine, One Baylor Plaza, Houston ,Texas 77030. Tel: 713 798 5862, email: [email protected]. Tel: 713 798 5860, email: [email protected] Running Head: Adrenergic activation of cardiac vagal neurons 1 bioRxiv preprint doi: https://doi.org/10.1101/276998; this version posted January 8, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Cholinergic vagal nerves projecting from neurons in the brainstem nucleus ambiguus (NAm) play a predominant role in cardiac parasympathetic pacemaking control. Central adrenergic signaling modulates the tone of this vagal output; however the exact excitability mechanisms are not fully understood. We investigated responses of NAm neurons to adrenergic agonists using in vitro mouse brainstem slices. Preganglionic NAm neurons were identified by Chat-tdtomato fluorescence in young adult transgenic mice and their cardiac projection confirmed by retrograde dye tracing. Juxtacellular recordings detected sparse or absent spontaneous action potentials (AP) in NAm neurons. However bath application of epinephrine or norepinephrine strongly and reversibly activated most NAm neurons regardless of their basal firing rate. -

Gene Therapy for Recurrent Laryngeal Nerve Injury
G C A T T A C G G C A T genes Review Gene Therapy for Recurrent Laryngeal Nerve Injury Koji Araki * ID , Hiroshi Suzuki, Kosuke Uno ID , Masayuki Tomifuji and Akihiro Shiotani Department of Otolaryngology-Head & Neck Surgery, National Defense Medical College, Saitama 3598513, Japan; [email protected] (H.S.); [email protected] (K.U.); [email protected] (M.T.); [email protected] (A.S.) * Correspondence: [email protected]; Tel.: +81-4-2995-1686 Received: 2 June 2018; Accepted: 20 June 2018; Published: 25 June 2018 Abstract: Recurrent laryngeal nerve (RLN) injury has considerable clinical implications, including voice and swallowing dysfunction, which may considerably impair the patient’s quality of life. Recovery of vocal fold movement is an essential novel treatment option for RLN injury. The potential of gene therapy for addressing this issue is highly promising. The target sites for RLN gene therapy are the central nervous system, nerve fibers, laryngeal muscles, and vocal cord mucosa. Gene transduction has been reported in each site using viral or non-viral methods. The major issues ensuing after RLN injury are loss of motoneurons in the nucleus ambiguus, degeneration and poor regeneration of nerve fibers and motor end plates, and laryngeal muscle atrophy. Gene therapy using neurotrophic factors has been assessed for most of these issues, and its efficacy has been reported. Another important matter for functional vocal fold movement recovery is misdirected regeneration, in which the wrong neurons may innervate other laryngeal muscles, where even if innervation is reestablished, proper motor function is not restored. -
Lecture 6: Cranial Nerves
Lecture 6: Cranial Nerves Objective: To understand the organization of cranial nerves with respect to their nuclei within the brain, their course through and exit from the brain, and their functional roles. Olfactory Eye Muscles 3, 4 &6 Cranial Nerves 1-7 I overview Table, Page 49 II Lecture notes Cranial Nerves and their Functions V Trigeminal VII Facial VIII IX X XII XI Cranial Nerves 8-12 Overview sternocephalic I. Factors Responsible for the Complex Internal Organization of the Brain Stem-> leads to altered location of cranial nerve nuclei in adult brain stem 1. Development of the Fourth Ventricle a. Medulla and Pons develop ventral to the 4th ventricle cerebellum b. Alar plate is displaced lateral to basal plate 4 Medulla Developing Neural Tube 2. Cranial nerve nuclei form discontinuous columns Rostral 12 SE Page 48 Notes 3. Some cranial nerve nuclei migrate from their primitive embryonic positions (e.g., nuclei of V and VII) Facial N. Factors responsible for the complex internal organization of the brainstem: 4) Special senses develop in association with the brain stem. Nuclei of special senses 5) Development of the cerebellum and its connections Cerebellum II. Cranial Nerve Nuclei: Nucleus = column of neuron cell bodies. Efferent nuclei are composed of cell bodies of alpha or gamma motor neurons (SE) or preganglionic parasympathetic neurons (VE). III. Motor Efferent Nuclei (Basal Plate Derivatives): 1. SE (Somatic Efferent) Nuclei: SE neurons form two longitudinally oriented but discontinuous columns of cell bodies in the brain stem. Neurons that comprise these columns are responsible for innervating all of the skeletal musculature of the head. -

Differentiation of the Bulbar Motor Nuclei and the Coincident Develop- Ment of Associated Root Fibers in the Rsbbit
DIFFERENTIATION OF THE BULBAR MOTOR NUCLEI AND THE COINCIDENT DEVELOP- MENT OF ASSOCIATED ROOT FIBERS IN THE RSBBIT DONALD L. KIMMEL Department of Anatomy, University of Michigan,’ Aim Arbor THIRTY-ONE FIGURES CONTENTS Introduction ........................................................ 83 Material and methods ................................................ 84 General survey of the literature ....................................... 85 Description of material studied ........................................ 86 Somatic efferent component. Its central and peripheral development .... 86 The hypoglossal ............................................. 86 The abdueens ............................................... 96 Visceral efferent component. Its central and peripheral development .... 103 The vago-accessory ........................................... 103 The glossopharyngeal ........................................ 124 The facial .................................................. 129 The trigemiiial .............................................. 137 Geiicrxldisrussion .................................................... 143 INTRODUCTION This present study concerns itself primarily with the onto- genetic development of the nuclear centers of bulbar cranial nerves in the rabbit and with the embryonic and adult distri- butions of their branches. Its purpose is to show that the central development proceeds stage for stage with the pro- A dissertation submitted in partial fulfillment of the requirements for the degree of doctor of philosophy -

Nuclear Architecture in the Medulla Oblongata of the Adult African Giant Pouched Rat (Cricetomys Gambianus, Waterhouse - 1840)
Int. J. Morphol., 29(2):382-388, 2011. Nuclear Architecture in the Medulla Oblongata of the Adult African Giant Pouched Rat (Cricetomys gambianus, Waterhouse - 1840) Arquitectura Nuclear en la Médula Oblonga de la Rata Gigante de Carillos Africana Adulta (Cricetomys gambianus, Waterhouse - 1840) *Ibe, C. S; *Onyeanusi, B. I.; *Hambolu, J. O. & **Ayo, J. O. IBE, C. S.; ONYEANUSI, B. I.; HAMBOLU, J. O. & AYO, J. O. Nuclear architecture in the medulla oblongata of the adult African giant pouched rat (Cricetomys gambianus, Waterhouse - 1840). Int. J. Morphol., 29(2):382-388, 2011. SUMMARY: The architecture of cranial and non-cranial nerve nuclei in the medulla oblongata of the African giant pouched rat was studied by means of light microscopy. Serial sections of the medulla oblongata, in coronal and saggital planes, were stained with the cresyl fast violet and silver stains, respectively. Sections in the saggital plane were used as a guide, while coronal sections were used to identify the nuclei in the rostrocaudal extent of the medulla oblongata. With the obex serving as the landmark, nuclei rostral and caudal to the obex were delineated. Cranial nerve nuclei whose architecture were defined were the motor nucleus of hypoglossal nerve, motor nucleus of vagus nerve, cochlear nucleus, vestibular nucleus and nucleus ambiguus, while non-cranial nerve nuclei identified were the olivary nucleus, solitary tract nucleus, gracile nucleus, cuneate nucleus, spinal nucleus of trigeminal nerve, motor nucleus of corpus trapezoideum, lateral nucleus of reticular formation and gigantocellular nucleus. The olivary nucleus was the most prominent nucleus, while the solitary tract nucleus was faint, and thus, less developed. -

Central Mechanisms of Cardiovascular Regulation During Exercise: Integrative Functions of the Nucleus of the Solitary Tract
J Phys Fitness Sports Med, 1(2): 253-261 (2012) JPFSM: Review Article Central mechanisms of cardiovascular regulation during exercise: Integrative functions of the nucleus of the solitary tract Hidefumi Waki Department of Physiology, Wakayama Medical University School of Medicine, 811-1 Kimiidera, Wakayama 641-8509, Japan Received: April 17, 2012 / Accepted: June 7, 2012 Abstract Generally, a single bout of exercise induces a moderate increase in arterial pressure (AP) with marked tachycardia as a result of sympathoexcitation which induces vasoconstriction in the major organs, but not in skeletal muscles, and activates heart function. In this review, the potential brain mechanisms underlying cardiovascular regulation during exercise are intro- duced, with a focus on the functions of the nucleus of the solitary tract (NTS), which is the cen- tral termination site of baroreceptor inputs. During a single bout of exercise, neuronal signals from the central command, mediated by the hypothalamus, as well as those from the muscle re- ceptors, are directly or indirectly projected to the NTS and rostral ventrolateral medulla (RVLM). The signals to the RVLM activate sympathetic premotor neurons that, in turn, induce pressor and tachycardiac responses. However, in the absence of resetting of the baroreceptor reflex to a higher pressure range, sympathoexcitation would be dampened and parasympathetic nerves would be excited by heightened levels of baroreceptor inputs, resulting in the attenuation of continuous increases in AP and heart rate. The GABAergic inter-neurons within the NTS may be involved in baroreceptor reflex resetting by limiting the degree of excitation of barosensi- tive NTS neurons, and thus are capable of ‘continuous’ increases in sympathetic nerve activ- ity. -

A Circumscribed Projection from the Nucleus of the Solitary Tract to The
The Journal of Neuroscience, May 1989, g(5): 1668-l 682 A Circumscribed Projection from the Nucleus of the Solitary Tract to the Nucleus Ambiguus in the Rat: Anatomical Evidence for Somatostatin-284mmunoreactive Interneurons Subserving Reflex Control of Esophageal Motility E. T. Cunningham, Jr.‘s21a and P. E. Sawchenko2 ‘Department of Neurosciences, University of California, San Diego, La Jolla, California 92093, and 2The Salk Institute for Biological Studies and The Clayton Foundation for Research-California Division, La Jolla, California 92037 Axonal transport and immunohistochemical methods were The nucleus of the solitary tract (NTS) occupies a pivotal po- used to investigate the anatomical and biochemical orga- sition in the central visceromotor system. Projections of the nization of projections from the nucleus of the solitary tract Vth, VIIth, IXth, and Xth cranial nerves terminate centrally (NTS) to the rostral, esophageal, part of the nucleus ambig- within the NTS, conveying sensoryinformation from the oral, uus (NA) in the rat. Discrete iontophoretic deposits of a ret- thoracic, and abdominal cavities. The NTS, in turn, projects to rogradely transported tracer, fluorogold, placed in the rostra1 virtually all levels of the neuraxis, so as to coordinate appro- NA labeled a column of cells within the NTS, termed the priate behavioral, neuroendocrine, and autonomic responses central part of the NTS (after Ross et al., 1985) situated just (seeSawchenko, 1983; Norgren, 1984, for reviews). medial to the solitary tract and extending -

The Brain Stem and Cerebellum: Part 1, the Medulla Introduction
The Brain Stem and Cerebellum: Part 1, the Medulla Brad Cole, MD Introduction The brain stem is a complex structure because of the multitude of various nuclei, pathways, and cranial nerves which are all in this relatively small area. It is an important structure clinically since there are a number of specific neurological syndromes which occur due to lesions in the brain stem. Because the cerebellum has so many connections with the brain stem, this will be covered in this section as well. In these lectures we will combine the normal anatomy with an introduction to the more common lesions within the brain stem. First some “big picture” points: Review books sometimes refer to the “rule of 4’s”. These are not entirely accurate (see footnotes) but I’ll share them here as a starting point: • There are 4 CNs in each section: 1 o Cranial nerves 3 and 4: Midbrain 2 o Cranial nerves 5-8: Pons o Cranial nerves 9-12: Medulla • CNs that divide evenly into 12 are found in the midline (3,4,6, and 12) • CNs that do not divide evenly into 12 are found more laterally (5,7,8,9,10 and 11)3 • 4 Midline pathways/structures: o Medial Longitudinal Fasciculus 4 o Motor tract of the Corticospinal Tract o Medial Lemniscus o Motor nuclei of CN 3,4,6, and 12 • 4 Sensory pathways are lateral: o Spinothalamic tract o Spinocerebellar tracts o Sympathetic descending first order pathway o Sensory CN nuclei Special thanks to Stephanie Hynes and Heidi Spady (both LLU class of 2020) for their artistic contributions to the brain stem handouts! 1 CNs 1-2 are CNS pathways and should not be included in this 2 CN 5 is also found in the medulla! 3 CN 7 is midline when it wraps around CN 6 in the pons 4 This pathway is lateral in the midbrain As we move from the medulla to the midbrain using the slides from the DeArmond atlas, you will need to be able to identify the structures that are labeled with *asterisks*. -
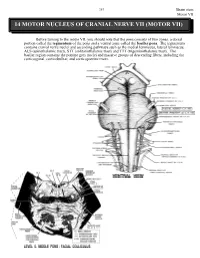
14 Motor Nucleus of Cranial Nerve Vii (Motor Vii)
263 Brain stem Motor VII 14 MOTOR NUCLEUS OF CRANIAL NERVE VII (MOTOR VII) Before turning to the motor VII, you should note that the pons consists of two zones, a dorsal portion called the tegmentum of the pons and a ventral zone called the basilar pons. The tegmentum contains cranial nerve nuclei and ascending pathways such as the medial lemniscus, lateral lemniscus, ALS (spinothalamic tract), STT (solitariothalamic tract) and TTT (trigeminothalamic tract). The basilar region contains the pontine grey nuclei and massive groups of descending fibers, including the corticospinal, corticobulbar, and corticopontine tracts. Brain stem 264 Motor VII The motor nucleus VII contains motor neurons (branchiomotor) that innervate the muscles of facial expression including the orbicularis oculi (CLOSES eyelid), the stapedius, the stylohyoid and the posterior belly of the digastric. Neurons comprising motor VII possess axons that pursue a rather circuitous route in order to exit the brain stem. Initially they pass dorsally and medially to loop over the abducens nucleus. The fibers then course ventrally and laterally to exit the brain stem. The bump in the floor of the fourth ventricle caused by the motor fibers of C.N. VII looping over the abducens nucleus is called the FACIAL COLLICULUS. A unilateral lesion interrupting the axons of C.N. VII results in the following: On the ipsilateral side, the forehead is immobile, the corner of the mouth sags, the nasolabial folds of the face are flattened, facial lines are lost, and saliva may drip from the corner of the mouth. The patient is unable to whistle or puff the cheek because the buccinator muscle is paralyzed. -
01 18-19-20-21 Cranial Nerve Nuclei-NOTES.Pdf
Cranial Nerve Nuclei Medical Neuroscience | Tutorial Notes Cranial Nerve Nuclei 1 MAP TO NEUROSCIENCE CORE CONCEPTS NCC1. The brain is the body's most complex organ. LEARNING OBJECTIVES After study of the assigned learning materials, the student will: 1. Identify the major subdivisions of the brainstem and spinal cord, as seen in representative transverse cross-sections. 2. Discuss the relationship between the cranial nerves and the corresponding cranial nerve nuclei. NARRATIVE by Leonard E. White and Nell B. Cant Department of Neurology Department of Neurobiology Duke University School of Medicine Duke Institute for Brain Sciences Introduction Of chief importance in understanding the organization of the brainstem is knowledge of what is localized in each embryological subdivision and in any transverse section. This is a significant challenge for every student of neuroanatomy and we will now turn our attention progressively to this challenge. You have already faced the first step toward competency with the essential knowledge: recognition of the external features of each brainstem subdivision, including the associated cranial nerves. After working through this tutorial, you should be able to recognize how the cranial nerves relate to gray matter structures in the brainstem that grew out the axons in the cranial nerves (motor axons) or receive synaptic input from ganglionic neurons associated with the nerves (sensory axons). Before proceeding, it will be worth reminding yourself of the basic layout of sensory and motor neurons in the brainstem