7: Approach to Glomerular Diseases
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Albumin in Health and Disease: Protein Metabolism and Function*
Article #2 CE An In-Depth Look: ALBUMIN IN HEALTH AND DISEASE Albumin in Health and Disease: Protein Metabolism and Function* Juliene L. Throop, VMD Marie E. Kerl, DVM, DACVIM (Small Animal Internal Medicine), DACVECC Leah A. Cohn, DVM, PhD, DACVIM (Small Animal Internal Medicine) University of Missouri-Columbia ABSTRACT: Albumin is a highly charged, 69,000 D protein with a similar amino acid sequence among many veterinary species. Albumin is the major contributor to colloid oncotic pressure and also serves as an important carrier protein. Its ability to modulate coagulation by preventing pathologic platelet aggrega- tion and augmenting antithrombin III is important in diseases that result in moderately to severely decreased serum albumin levels. Synthesis is primar- ily influenced by oncotic pressure, but inflammation, hormone status, and nutrition impact the synthetic rate as well. Albumin degradation is a poorly understood process that does not appear to be selective for older mole- cules.The clinical consequences of hypoalbuminemia reflect the varied func- tions of the ubiquitous albumin protein molecule. he albumin molecule has several characteristics that make it a unique protein. Most veterinarians are aware of the importance of this molecule in maintain- Ting colloid oncotic pressure, but albumin has many other less commonly recog- nized functions as well. The clinical consequences of hypoalbuminemia are reflections of the many functions albumin fulfills. Understanding the functions, synthesis, and *A companion article on degradation of the albumin molecule can improve understanding of the causes, conse- causes and treatment of quences, and treatment of hypoalbuminemia. hypoalbuminemia appears on page 940. STRUCTURE Email comments/questions to Albumin has a molecular weight of approximately 69,000 D, with minor variations [email protected], among species. -

Renal Sodium Handling in Minimal Change Nephrotic Syndrome
Arch Dis Child: first published as 10.1136/adc.59.9.825 on 1 September 1984. Downloaded from Archives of Disease in Childhood, 1984, 59, 825-830 Renal sodium handling in minimal change nephrotic syndrome A-B BOHLIN AND U BERG Department of Paediatrics, Karolinska Institute, Huddinge Hospital, Stockholm, Sweden SUMMARY Renal sodium handling was studied in 23 children at three different stages of the minimal change nephrotic syndrome-the oedema forming state, proteinuric steady state, and remission. Clearances of inulin and para-aminohippuric acid and urinary sodium excretion were determined basally, after intravenous infusion of isotonic saline and hyperoncotic albumin, and after furosemide injection. Absolute and fractional basal sodium excretion were significantly lower in oedema forming patients than in proteinuric patients in steady state, and non-proteinuric patients. In contrast to proteinuric patients in steady state and non-proteinuric patients, the oedema forming patients failed to respond to isotonic saline infusion with increased sodium excretion. After diuretic blockade with furosemide, the fractional sodium excretion of the oedema forming patients increased to values no different from those of the non-proteinuric patients, whereas the fractional sodium excretion of the steady state patients increased to significantly higher values. The plasma aldosterone concentration was within normal limits in 11 of 14 proteinuric patients, and did not correlate with the basal sodium excretion. Thus, sodium copyright. retention in the minimal change nephrotic syndrome was found only in oedema forming patients, and since this is not related to the plasma aldosterone concentration it may be caused by an intrarenal mechanism, probably sited in distal parts of the nephron. -

PATHOLOGY of the RENAL SYSTEM”, I Hope You Guys Like It
ﺑﺴﻢ اﷲ اﻟﺮﺣﻤﻦ اﻟﺮﺣﯿﻢ ھﺬه اﻟﻤﺬﻛﺮة ﻋﺒﺎرة ﻋﻦ إﻋﺎدة ﺗﻨﺴﯿﻖ وإﺿﺎﻓﺔ ﻧﻮﺗﺎت وﻣﻮاﺿﯿﻊ ﻟﻤﺬﻛﺮة زﻣﻼﺋﻨﺎ ﻣﻦ اﻟﺪﻓﻌﺔ اﻟﺴﺎﺑﻘﺔ ٤٢٧ اﻷﻋﺰاء.. ﻟﺘﺘﻮاﻓﻖ ﻣﻊ اﻟﻤﻨﮭﺞ اﻟﻤﻘﺮر ﻣﻦ اﻟﻘﺴﻢ ﺣﺮﺻﻨﺎ ﻓﯿﮭﺎ ﻋﻠﻰ إﻋﺎدة ﺻﯿﺎﻏﺔ ﻛﺜﯿﺮ ﻣﻦ اﻟﺠﻤﻞ ﻟﺘﻜﻮن ﺳﮭﻠﺔ اﻟﻔﮭﻢ وﺳﻠﺴﺔ إن ﺷﺎء اﷲ.. وﺿﻔﻨﺎ ﺑﻌﺾ اﻟﻨﻮﺗﺎت اﻟﻤﮭﻤﺔ وأﺿﻔﻨﺎ ﻣﻮاﺿﯿﻊ ﻣﻮﺟﻮدة ﺑﺎﻟـ curriculum ﺗﻌﺪﯾﻞ ٤٢٨ ﻋﻠﻰ اﻟﻤﺬﻛﺮة ﺑﻮاﺳﻄﺔ اﺧﻮاﻧﻜﻢ: ﻓﺎرس اﻟﻌﺒﺪي ﺑﻼل ﻣﺮوة ﻣﺤﻤﺪ اﻟﺼﻮﯾﺎن أﺣﻤﺪ اﻟﺴﯿﺪ ﺣﺴﻦ اﻟﻌﻨﺰي ﻧﺘﻤﻨﻰ ﻣﻨﮭﺎ اﻟﻔﺎﺋﺪة ﻗﺪر اﻟﻤﺴﺘﻄﺎع، وﻻ ﺗﻨﺴﻮﻧﺎ ﻣﻦ دﻋﻮاﺗﻜﻢ ! 2 After hours, or maybe days, of working hard, WE “THE PATHOLOGY TEAM” are proud to present “PATHOLOGY OF THE RENAL SYSTEM”, I hope you guys like it . Plz give us your prayers. Credits: 1st part = written by Assem “ THe AWesOme” KAlAnTAn revised by A.Z.K 2nd part = written by TMA revised by A.Z.K د.ﺧﺎﻟﺪ اﻟﻘﺮﻧﻲ 3rd part = written by Abo Malik revised by 4th part = written by A.Z.K revised by Assem “ THe AWesOme” KAlAnTAn 5th part = written by The Dude revised by TMA figures were provided by A.Z.K Page styling and figure embedding by: If u find any error, or u want to share any idea then plz, feel free to msg me [email protected] 3 Table of Contents Topic page THE NEPHROTIC SYNDROME 4 Minimal Change Disease 5 MEMBRANOUS GLOMERULONEPHRITIS 7 FOCAL SEGMENTAL GLOMERULOSCLEROSIS 9 MEMBRANOPROLIFERATIVE GLOMERULONEPHRITIS 11 DIABETIC NEPHROPATHY (new) 14 NEPHRITIC SYNDROME 18 Acute Post-infectious GN 19 IgA Nephropathy (Berger Disease) 20 Crescentic GN 22 Chronic GN 24 SLE Nephropathy (new) 26 Allograft rejection of the transplanted kidney (new) 27 Urinary Tract OBSTRUCTION, 28 RENAL STONES 23 HYDRONEPHROSIS -

Annotations Prognosis for Vesicoureteric Reflux
Arch Dis Child 1999;81:287–294 287 Arch Dis Child: first published as 10.1136/adc.81.4.287 on 1 October 1999. Downloaded from The Journal of the Royal College of Paediatrics and Child Health Annotations Prognosis for vesicoureteric reflux The prevalence of vesicoureteric reflux (VUR) has been to disentangle in this group of patients. The development estimated to be 2% of the child population.1 In children with of proteinuria is indicative of progressive glomerulosclero- VUR demonstrated on micturating cystourethrography sis and is a bad prognostic feature particularly when the there is a tendency for the grade of VUR to improve or for patient also has hypertension. VUR to disappear with time and with increasing age.23VUR has been identified as a risk factor for the development of Historical perspective urinary tract infections (UTI) and is present in a third of A review of literature in the preantibiotic era suggests that young children presenting with this problem. In addition, it chronic pyelonephritis was a very serious condition in chil- is a risk factor for renal scarring, otherwise called reflux dren and adults. Weiss and Parker described a series of nephropathy.45 VUR is also associated with renal dysplasia postmortem cases16: antecedent clinical features included and other developmental abnormalities of the urinary tract.6 recurrent fevers, presumably due to persistent untreated There is now abundant evidence for inheritance by an auto- infection, anaemia, hypertension, growth failure, and preg- somal dominant mechanism.7 nancy complications. There is evidence for a falling preva- lence of this condition, which is probably due to a true reduction of reflux nephropathy because of modern medi- Pathogenesis of reflux nephropathy cal care, particularly the treatment of acute pyelonephritis Studies have suggested that reflux nephropathy develops with antibiotics; alternatively the decline may represent following UTI in very early childhood or infancy.8 New changing fashions in disease classification. -

Glomerulosclerosis in Reflux Nephropathy
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Kidney International, Vol. 21(1982), pp. 528—534 NEPHROLOGY FORUM Glomeruloscierosis in reflux nephropathy Principal discussant: RAMzI S. COTRAN Department of Pathology, Brigham and Women's Hospital, Boston, Massachusetts penis, testes, and urethral meatus were normal. The prostate was of normal size. Editors The BUN was 14 mg/dl; creatinine, 1.3 mg/dl (creatinine clearance, 113 mI/mm); blood chemistries were normal; the blood glucose was 93 JORDANJ. COHEN mg/dl; and the complement profile was normal. Serum protein was 7.5 JOHN 1. HARRINGTON gIdI with 4.3 g/dl albumin. Urinalysis showed a pH of 5; a specific JEROME P.KASSIRER gravity of 1.015; 4+ protein, no cells, and no bacteria. Urine culture was sterile. The 24-hour urine protein excretion was 2.4 g. Editor Chest x-ray showed borderline cardiomegaly with clear lungs. An Managing intravenous pyelogram revealed bilateral coarse scarring with caliecta- CHERYL J. ZUSMAN sis. The right kidney was smaller than the left. There was moderate ureterectasia extending down to the ureterovesical junction. A voiding cystourethrogram revealed a large-capacity bladder; the patient had no MichaelReese Hospital and Medical Center urge to void after almost 500 ml of contrast material was instilled. Bilateral reflux was greater and persistent on the left and was intermit- University of Chicago, tent on the right. The left ureter was dilated and tortuous. A left Pritzker School of Medicine ureterocele and right bladder diverticulum were visualized. and A biopsy of the left kidney showed focal scarring with interstitial New England Medical Center fibrosis and chronic inflammation, tubular atrophy, and dilation. -

Predictors of Vesicoureteral Reflux in the Pretransplant Evaluation of Patients with End-Stage Renal Disease
DOI: 10.14744/scie.2018.63935 Original Article South. Clin. Ist. Euras. 2018;29(3):176-179 Predictors of Vesicoureteral Reflux in the Pretransplant Evaluation of Patients with End-Stage Renal Disease Ergün Parmaksız, Meral Meşe, Zuhal Doğu, Zerrin Bicik Bahçebaşı ABSTRACT Objective: Voiding cystourethrography (VCUG) is widely performed in the pretransplant Department of Nephrology, evaluation of patients with a history of urological disorders to detect vesicoureteral reflux University of Health Sciences (VUR). The aim of this study was to evaluate the relationship between the primary etiology Kartal Dr. Lütfi Kırdar Training and Research Hospital, İstanbul, Turkey of end-stage renal disease (ESRD) and the prevalence of VUR, thereby determining the ne- cessity for VCUG in pretransplant patients. Submitted: 10.05.2018 Accepted: 27.08.2018 Methods: A total of 319 pretransplant cases that underwent VCUG were retrospectively reviewed. Correspondence: Ergün Parmaksız, SBÜ Kartal Dr. Lütfi Kırdar Results: VCUG revealed VUR in 53 (16.6%) cases. VUR was left-sided in 21 (41.2%), right- Eğitim ve Araştırma Hastanesi, Nefroloji Kliniği, İstanbul, Turkey sided in 18 (35.3%), and bilateral in 12 (3.8%), and grade 1 in 10 (19.6%), grade 2 in 19 E-mail: [email protected] (37.3%), grade 3 in 20 (39.2%), and grade 4 in 2 (3.9%). The etiology of ESRD was hyperten- sion in 125 (39.2%), diabetes mellitus (DM) in 46 (14.4%), polycystic kidney disease (PKD) in 21 (6.6%), amyloidosis in 16 (5%), VUR in 11 (3.4%), and glomerulonephritis (GN) in 11 (3.4%). The incidence of VUR was significantly higher in female patients. -

Determinants of Glomerular Filtration in Experimental Glomerulonephritis in the Rat
Determinants of glomerular filtration in experimental glomerulonephritis in the rat. D A Maddox, … , T M Daugharty, B M Brenner J Clin Invest. 1975;55(2):305-318. https://doi.org/10.1172/JCI107934. Research Article Pressures and flows were measured in surface glomerular capillaries, efferent arterioles, and proximal tubules of 22 Wistar rats in the early autologous phase of nephrotoxic serum nephritis (NSN). Linear deposits of rabbit and rat IgG and C3 component of complement were demonstrated in glomerular capillary walls by immunofluorescence microscopy. Light microscopy revealed diffuse proliferative glomerulonephritis, and proteinuria was present. Although whole kidney and single nephron glomerular filtration rate (GFR) in NSN (0.8 plus or minus 0.04 SE2 ml/min and 2 plus or minus 2 nl/min, respectively) remained unchanged from values in 16 weight-matched NORMAL HYDROPENIC control rats (0.8 plus or minus 0.08 and 28 plus or minus 2), important alterations in glomerular dynamics were noted. Mean transcapillary hydraulic pressure difference (deltaP) averaged 41 plus or minus 1 mm Hg in NSN versus 32 plus or minus 1 in controls (P LESS THAN 0.005). Oncotic pressures at the afferent (piA) end of the glomerular capillary were similar in both groups ( 16 mm /g) but increased much less by the efferent end (piE) in NSN (to 29 plus or minus 1 mm Hg) than in controls (33 plus or minus 1, P less than 0.025). Hence, equality between deltaP and piE, denoting filtration pressure equilibrium, obtained in control but not in NSN rats. While glomerular plasma flow rate was slightly higher […] Find the latest version: https://jci.me/107934/pdf Determinants of Glomerular Filtration in Experimental Glomerulonephritis in the Rat D. -

Glomerulonephritis Management in General Practice
Renal disease • THEME Glomerulonephritis Management in general practice Nicole M Isbel MBBS, FRACP, is Consultant Nephrologist, Princess Alexandra lomerular disease remains an important cause Hospital, Brisbane, BACKGROUND Glomerulonephritis (GN) is an G and Senior Lecturer in important cause of both acute and chronic kidney of renal impairment (and is the commonest cause Medicine, University disease, however the diagnosis can be difficult of end stage kidney disease [ESKD] in Australia).1 of Queensland. nikky_ due to the variability of presenting features. Early diagnosis is essential as intervention can make [email protected] a significant impact on improving patient outcomes. OBJECTIVE This article aims to develop However, presentation can be variable – from indolent a structured approach to the investigation of patients with markers of kidney disease, and and asymptomatic to explosive with rapid loss of kidney promote the recognition of patients who need function. Pathology may be localised to the kidney or further assessment. Consideration is given to the part of a systemic illness. Therefore diagnosis involves importance of general measures required in the a systematic approach using a combination of clinical care of patients with GN. features, directed laboratory and radiological testing, DISCUSSION Glomerulonephritis is not an and in many (but not all) cases, a kidney biopsy to everyday presentation, however recognition establish the histological diagnosis. Management of and appropriate management is important to glomerulonephritis (GN) involves specific therapies prevent loss of kidney function. Disease specific directed at the underlying, often immunological cause treatment of GN may require specialist care, of the disease and more general strategies aimed at however much of the management involves delaying progression of kidney impairment. -

Glomerular Filtration I DR.CHARUSHILA RUKADIKAR Assistant Professor Physiology GFR 1
Glomerular filtration I DR.CHARUSHILA RUKADIKAR Assistant Professor Physiology GFR 1. Definition 2. Normal value 3. Variation 4. Calculation (different pressures acting on glomerular membrane) 5. Factors affecting GFR 6. Regulation of GFR 7. Measurement of GFR QUESTIONS LONG QUESTION 1. GFR 2. RENIN ANGIOTENSIN SYSTEM SHORT NOTE 1. DYNAMICS OF GFR 2. FILTRATION FRACTION 3. ANGIOTENSIN II 4. FACTORS AFFECTING GLOMERULAR FILTRATION RATE 5. REGULATION OF GFR 6. RENAL CLEARANCE TEST 7. MEASUREMENT OF GFR Collecting duct epithelium P Cells – Tall, predominant, have few organelles, Na reabsorption & vasopressin stimulated water reabsorption I cells- CT and DCT, less, having more cell organelles, Acid secretion and HCO3 transport CHARACTERISTICS OF RENAL BLOOD FLOW 600-1200 ml/min (high) AV O2 difference low (1.5 mL/dL) During exercise increases 1.5 times Low basal tone, not altered in denervated / innervated kidney VO2 in kidneys is directly proportional to RBF, Na reabsorption & GFR Not homogenous flow, cortex more & medulla less Vasa recta hairpin bend like structure, hyperosmolarity inner medulla Transplanted kidney- cortical blood flow show autoregulation & medullary blood flow don’t show autoregulation, so no TGF mechanism Neurogenic vasodilation not exist 20% of resting cardiac output, while the two kidneys make < 0.5% of total body weight. Excretory function rather than its metabolic requirement. Remarkable constancy due to autoregulation. Processes concerned with urine formation. 1. Glomerular filtration, 2. Tubular reabsorption and 3. Tubular secretion. • Filtration Fluid is squeezed out of glomerular capillary bed • Reabsorption Most nutrients, water and essential ions are returned to blood of peritubular capillaries • Secretion Moves additional undesirable molecules into tubule from blood of peritubular capillaries Glomerular filtration Glomerular filtration refers to process of ultrafiltration of plasma from glomerular capillaries into the Bowman’s capsule. -

Significance of Protein in Edema Fluids I C
i Significance of Protein in Edema Fluids I C. L. Witte, M. H. "f itte, A. E. Dumont Departments of Surgery, University of Arizona College of Medicine, Tucson, Arizona and "'ewYorkl.Jniversity_School of Medicine, New York, N. Y. Lymphology 4 (1971), 29-31 The partition of extracellular fluid between plasma and tissues is largely regulated by the balance of hydrostatic an~ colloid osmotic (oncotic) pressure gradients across capillary membranes. Normally ~ small excess of tissue fluid forms, enters lymphatics and returns to the bloodstream. When an imbalance develops between the rate of lymph production and the rate of its return to the systemic venous circulation, edema.or effusion appear. Accumulation of edema, however, is not uniform in amount and composition throughout the body and depend, on the forces governing capillary filtration, regional differences in capillary permeabiFty, lymph flow and the specific factor tending to per petuate the edema. Obstruction td lymph return or disruption of capillary integrity (e.g. I infiltrating cancer or inflammation) promote effusion or edema high in protein content (so-called "exudate"). On the other hand, increased hydrostatic pressure in semiperme able capillaries promotes excess lymph and ultimately edema or effusion low in protein content (so-called "transudate"). iThe hepatic sinusoid is unique in its free permeability to plasma protein. Hepatic venous outflow obstruction, accordingly, produces excess hepatic and thoracic duct lymphi and eventually ascitic fluid, high in protein. In his monograph on The Fluids of the Body (1), Starling reasoned that increased capillary hydrostatic pressure Jas initially off set by a fall in tissue oncotic pressure. As the gradient in hydrostatic pressure rises in semipermeable capillary beds a progres sively more dilute capillary filtrate forms in tissues. -
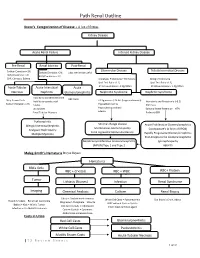
Path Renal Outline
Path Renal Outline Krane’s Categorization of Disease + A lot of Extras Kidney Disease Acute Renal Failure Intrinsic Kidney Disease Pre‐Renal Renal Intrinsic Post‐Renal Sodium Excretion <1% Glomerular Disease Tubulointerstitial Disease Sodium Excretion < 1% Sodium Excretion >2% Labs aren’t that useful BUN/Creatinine > 20 BUN/Creatinine < 10 CHF, Cirrhosis, Edema Urinalysis: Proteinuria + Hematuria Benign Proteinuria Spot Test Ratio >1.5, Spot Test Ratio <1.5, Acute Tubular Acute Interstitial Acute 24 Urine contains > 2.0g/24hrs 24 Urine contains < 1.0g/24hrs Necrosis Nephritis Glomerulonephritis Nephrotic Syndrome Nephritic Syndrome Inability to concentrate Urine RBC Casts Dirty Brown Casts Inability to secrete acid >3.5g protein / 24 hrs (huge proteinuria) Hematuria and Proteinuria (<3.5) Sodium Excretion >2% Edema Hypoalbuminemia RBC Casts Hypercholesterolemia Leukocytes Salt and Water Retention = HTN Focal Tubular Necrosis Edema Reduced GFR Pyelonephritis Minimal change disease Allergic Interstitial Nephritis Acute Proliferative Glomerulonephritis Membranous Glomerulopathy Analgesic Nephropathy Goodpasture’s (a form of RPGN) Focal segmental Glomerulosclerosis Rapidly Progressive Glomerulonephritis Multiple Myeloma Post‐Streptococcal Glomerulonephritis Membranoproliferative Glomerulonephritis IgA nephropathy (MPGN) Type 1 and Type 2 Alport’s Meleg‐Smith’s Hematuria Break Down Hematuria RBCs Only RBC + Crystals RBC + WBC RBC+ Protein Tumor Lithiasis (Stones) Infection Renal Syndrome Imaging Chemical Analysis Culture Renal Biopsy Calcium -

Nephrology Clinical Undergraduate Training
School of Medicine Nephrology clinical undergraduate training All care is taken to ensure that the information in this handbook is correct at the time of going to print. Handbook Version 5.0 Date of Origin: March 2019 Contents OBJECTIVES OF ATTACHMENT ................................................................................................... 3 TEACHING STRUCTURE: 3RD MEDICAL YEAR CLINICAL MEDICINE ATTACHMENTS .......................... 3 PROPOSED ATTACHMENT TIMETABLE: ................................................................................................ 5 READING LIST AND WEBSITES ............................................................................................................. 6 2 3rd Medical Year SPECIALTY: Nephrology - www.tcd.ie/medicine/thkc/education/ CONSULTANT: Prof G. Mellotte, Prof C Wall, Dr P Lavin, Dr B Griffin, Prof M Little HOSPITAL: Trinity Health Kidney Centre, Tallaght Hospital YEAR OF COURSE: 3 OBJECTIVES OF ATTACHMENT During this attachment, a student is expected to understand: - The clinical presentation of renal disease, e.g., proteinuria, hypertension, haematuria and uraemia. - Normal regulation of body water and sodium by the RAAS and ADH and how abnormalities give rise to changes in water and sodium homeostasis - Normal values of electrolytes in blood and urine and the clinical sequelae of a derangement in these. - A basic understanding of the following conditions: a. Acute kidney injury (pre-renal, post renal or intrinsic renal) b. Chronic kidney disease, focusing on diabetic nephropathy c. Glomerulonephritis: nephrotic / nephritic syndrome d. Myeloma and the kidney - The management of acute and chronic renal failure, including preparation for dialysis. - Impact of renal failure on drug handling. Be able to: - Take a full and appropriate current and past medical history. - Construct a synopsis or problem list based on the clinical assessment of a patient - Discuss the range of clinical investigations available and understand how they may be used to inform the differential diagnosis.