Targeting Oncoimmune Drivers of Cancer Metastasis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Precision Medicine for Human Cancers with Notch Signaling Dysregulation (Review)
INTERNATIONAL JOURNAL OF MOleCular meDICine 45: 279-297, 2020 Precision medicine for human cancers with Notch signaling dysregulation (Review) MASUKO KATOH1 and MASARU KATOH2 1M & M PrecMed, Tokyo 113-0033; 2Department of Omics Network, National Cancer Center, Tokyo 104-0045, Japan Received September 16, 2019; Accepted November 20, 2019 DOI: 10.3892/ijmm.2019.4418 Abstract. NOTCH1, NOTCH2, NOTCH3 and NOTCH4 are conjugate (ADC) Rova-T, and DLL3-targeting chimeric antigen transmembrane receptors that transduce juxtacrine signals of receptor‑modified T cells (CAR‑Ts), AMG 119, are promising the delta-like canonical Notch ligand (DLL)1, DLL3, DLL4, anti-cancer therapeutics, as are other ADCs or CAR-Ts targeting jagged canonical Notch ligand (JAG)1 and JAG2. Canonical tumor necrosis factor receptor superfamily member 17, Notch signaling activates the transcription of BMI1 proto-onco- CD19, CD22, CD30, CD79B, CD205, Claudin 18.2, fibro- gene polycomb ring finger, cyclin D1, CD44, cyclin dependent blast growth factor receptor (FGFR)2, FGFR3, receptor-type kinase inhibitor 1A, hes family bHLH transcription factor 1, tyrosine-protein kinase FLT3, HER2, hepatocyte growth factor hes related family bHLH transcription factor with YRPW receptor, NECTIN4, inactive tyrosine-protein kinase 7, inac- motif 1, MYC, NOTCH3, RE1 silencing transcription factor and tive tyrosine-protein kinase transmembrane receptor ROR1 transcription factor 7 in a cellular context-dependent manner, and tumor-associated calcium signal transducer 2. ADCs and while non-canonical Notch signaling activates NF-κB and Rac CAR-Ts could alter the therapeutic framework for refractory family small GTPase 1. Notch signaling is aberrantly activated cancers, especially diffuse-type gastric cancer, ovarian cancer in breast cancer, non-small-cell lung cancer and hematological and pancreatic cancer with peritoneal dissemination. -

Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse
Welcome to More Choice CD Marker Handbook For more information, please visit: Human bdbiosciences.com/eu/go/humancdmarkers Mouse bdbiosciences.com/eu/go/mousecdmarkers Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse CD3 CD3 CD (cluster of differentiation) molecules are cell surface markers T Cell CD4 CD4 useful for the identification and characterization of leukocytes. The CD CD8 CD8 nomenclature was developed and is maintained through the HLDA (Human Leukocyte Differentiation Antigens) workshop started in 1982. CD45R/B220 CD19 CD19 The goal is to provide standardization of monoclonal antibodies to B Cell CD20 CD22 (B cell activation marker) human antigens across laboratories. To characterize or “workshop” the antibodies, multiple laboratories carry out blind analyses of antibodies. These results independently validate antibody specificity. CD11c CD11c Dendritic Cell CD123 CD123 While the CD nomenclature has been developed for use with human antigens, it is applied to corresponding mouse antigens as well as antigens from other species. However, the mouse and other species NK Cell CD56 CD335 (NKp46) antibodies are not tested by HLDA. Human CD markers were reviewed by the HLDA. New CD markers Stem Cell/ CD34 CD34 were established at the HLDA9 meeting held in Barcelona in 2010. For Precursor hematopoetic stem cell only hematopoetic stem cell only additional information and CD markers please visit www.hcdm.org. Macrophage/ CD14 CD11b/ Mac-1 Monocyte CD33 Ly-71 (F4/80) CD66b Granulocyte CD66b Gr-1/Ly6G Ly6C CD41 CD41 CD61 (Integrin b3) CD61 Platelet CD9 CD62 CD62P (activated platelets) CD235a CD235a Erythrocyte Ter-119 CD146 MECA-32 CD106 CD146 Endothelial Cell CD31 CD62E (activated endothelial cells) Epithelial Cell CD236 CD326 (EPCAM1) For Research Use Only. -

Predictive QSAR Tools to Aid in Early Process Development of Monoclonal Antibodies
Predictive QSAR tools to aid in early process development of monoclonal antibodies John Micael Andreas Karlberg Published work submitted to Newcastle University for the degree of Doctor of Philosophy in the School of Engineering November 2019 Abstract Monoclonal antibodies (mAbs) have become one of the fastest growing markets for diagnostic and therapeutic treatments over the last 30 years with a global sales revenue around $89 billion reported in 2017. A popular framework widely used in pharmaceutical industries for designing manufacturing processes for mAbs is Quality by Design (QbD) due to providing a structured and systematic approach in investigation and screening process parameters that might influence the product quality. However, due to the large number of product quality attributes (CQAs) and process parameters that exist in an mAb process platform, extensive investigation is needed to characterise their impact on the product quality which makes the process development costly and time consuming. There is thus an urgent need for methods and tools that can be used for early risk-based selection of critical product properties and process factors to reduce the number of potential factors that have to be investigated, thereby aiding in speeding up the process development and reduce costs. In this study, a framework for predictive model development based on Quantitative Structure- Activity Relationship (QSAR) modelling was developed to link structural features and properties of mAbs to Hydrophobic Interaction Chromatography (HIC) retention times and expressed mAb yield from HEK cells. Model development was based on a structured approach for incremental model refinement and evaluation that aided in increasing model performance until becoming acceptable in accordance to the OECD guidelines for QSAR models. -

SPECIALTY MEDICATIONS Available Through Accredo Health Group, Inc., Medco’S Specialty Pharmacy Call Toll-Free (800) 803-2523, 8:00 A.M
SPECIALTY MEDICATIONS available through Accredo Health Group, Inc., Medco’s specialty pharmacy Call toll-free (800) 803-2523, 8:00 a.m. to 8:00 p.m., eastern time, Monday through Friday, to confirm that your medication is covered. Effective as of July 1, 2011 Abraxane® (paclitaxel protein-bound particles) Berinert® (C 1 esterase inhibitor [human])* (PA) (QD) Actemra ™ (tocilizumab) (PA) Betaseron® (interferon beta-1b) (PA) Actimmune® (interferon gamma-1b) (PA) Botox® (botulinum toxin type A) (PA) Adagen® (pegademase bovine) Carbaglu ™ (carglumic acid) Adcirca® (tadalafil) (ST) (QD) Carimune® NF (immune globulin intravenous [human]) (PA) Advate® (antihemophilic factor [recombinant]) (CPA) Cerezyme® (imiglucerase) (CPA) (ST) Afinitor® (everolimus) (PA) (QD) Cimzia® (certolizumab pegol) (ST) Aldurazyme® (laronidase) (CPA) Copaxone® (glatiramer acetate) (PA) Alphanate® (antihemophilic factor [human]) (CPA) Copegus® (ribavirin) (ST) AlphaNine® SD (coagulation factor IX [human]) (CPA) Corifact® (factor XIII [human]) (CPA) Amevive® (alefacept) (PA) Cystadane® (betaine) Ampyra ™ (dalfampridine) (PA) CytoGam® (cytomegalovirus immune globulin Apokyn® (apomorphine hydrochloride) (PA) (QD) intravenous [human])* (CPA) Aralast® (alpha[1]-proteinase inhibitor [human]) Cytovene® IV (ganciclovir sodium)* Aranesp® (darbepoetin alfa) (PA) Dacogen® (decitabine) Arcalyst® (rilonacept) (PA) (QD) Dysport® (abobotulinumtoxinA) (PA) Arixtra® (fondaparinux sodium)* Egrifta ™ (tesamorelin) (PA) Arranon® (nelarabine) Elaprase® (idursulfase) (CPA) Arzerra® (ofatumumab) -

Chronic Myeloid Leukemia: Mechanisms of Blastic Transformation
Chronic myeloid leukemia: mechanisms of blastic transformation Danilo Perrotti, … , John Goldman, Tomasz Skorski J Clin Invest. 2010;120(7):2254-2264. https://doi.org/10.1172/JCI41246. Science in Medicine The BCR-ABL1 oncoprotein transforms pluripotent HSCs and initiates chronic myeloid leukemia (CML). Patients with early phase (also known as chronic phase [CP]) disease usually respond to treatment with ABL tyrosine kinase inhibitors (TKIs), although some patients who respond initially later become resistant. In most patients, TKIs reduce the leukemia cell load substantially, but the cells from which the leukemia cells are derived during CP (so-called leukemia stem cells [LSCs]) are intrinsically insensitive to TKIs and survive long term. LSCs or their progeny can acquire additional genetic and/or epigenetic changes that cause the leukemia to transform from CP to a more advanced phase, which has been subclassified as either accelerated phase or blastic phase disease. The latter responds poorly to treatment and is usually fatal. Here, we discuss what is known about the molecular mechanisms leading to blastic transformation of CML and propose some novel therapeutic approaches. Find the latest version: https://jci.me/41246/pdf Science in medicine Chronic myeloid leukemia: mechanisms of blastic transformation Danilo Perrotti,1 Catriona Jamieson,2 John Goldman,3 and Tomasz Skorski4 1Department of Molecular Virology, Immunology and Medical Genetics and Comprehensive Cancer Center, The Ohio State University, Columbus, Ohio, USA. 2Division of Hematology-Oncology, Department of Internal Medicine, University of California at San Diego, La Jolla, California, USA. 3Department of Haematology, Imperial College at Hammersmith Hospital, London, United Kingdom. 4Department of Microbiology and Immunology, Temple University, Philadelphia, Pennsylvania, USA. -

Classification Decisions Taken by the Harmonized System Committee from the 47Th to 60Th Sessions (2011
CLASSIFICATION DECISIONS TAKEN BY THE HARMONIZED SYSTEM COMMITTEE FROM THE 47TH TO 60TH SESSIONS (2011 - 2018) WORLD CUSTOMS ORGANIZATION Rue du Marché 30 B-1210 Brussels Belgium November 2011 Copyright © 2011 World Customs Organization. All rights reserved. Requests and inquiries concerning translation, reproduction and adaptation rights should be addressed to [email protected]. D/2011/0448/25 The following list contains the classification decisions (other than those subject to a reservation) taken by the Harmonized System Committee ( 47th Session – March 2011) on specific products, together with their related Harmonized System code numbers and, in certain cases, the classification rationale. Advice Parties seeking to import or export merchandise covered by a decision are advised to verify the implementation of the decision by the importing or exporting country, as the case may be. HS codes Classification No Product description Classification considered rationale 1. Preparation, in the form of a powder, consisting of 92 % sugar, 6 % 2106.90 GRIs 1 and 6 black currant powder, anticaking agent, citric acid and black currant flavouring, put up for retail sale in 32-gram sachets, intended to be consumed as a beverage after mixing with hot water. 2. Vanutide cridificar (INN List 100). 3002.20 3. Certain INN products. Chapters 28, 29 (See “INN List 101” at the end of this publication.) and 30 4. Certain INN products. Chapters 13, 29 (See “INN List 102” at the end of this publication.) and 30 5. Certain INN products. Chapters 28, 29, (See “INN List 103” at the end of this publication.) 30, 35 and 39 6. Re-classification of INN products. -

Tanibirumab (CUI C3490677) Add to Cart
5/17/2018 NCI Metathesaurus Contains Exact Match Begins With Name Code Property Relationship Source ALL Advanced Search NCIm Version: 201706 Version 2.8 (using LexEVS 6.5) Home | NCIt Hierarchy | Sources | Help Suggest changes to this concept Tanibirumab (CUI C3490677) Add to Cart Table of Contents Terms & Properties Synonym Details Relationships By Source Terms & Properties Concept Unique Identifier (CUI): C3490677 NCI Thesaurus Code: C102877 (see NCI Thesaurus info) Semantic Type: Immunologic Factor Semantic Type: Amino Acid, Peptide, or Protein Semantic Type: Pharmacologic Substance NCIt Definition: A fully human monoclonal antibody targeting the vascular endothelial growth factor receptor 2 (VEGFR2), with potential antiangiogenic activity. Upon administration, tanibirumab specifically binds to VEGFR2, thereby preventing the binding of its ligand VEGF. This may result in the inhibition of tumor angiogenesis and a decrease in tumor nutrient supply. VEGFR2 is a pro-angiogenic growth factor receptor tyrosine kinase expressed by endothelial cells, while VEGF is overexpressed in many tumors and is correlated to tumor progression. PDQ Definition: A fully human monoclonal antibody targeting the vascular endothelial growth factor receptor 2 (VEGFR2), with potential antiangiogenic activity. Upon administration, tanibirumab specifically binds to VEGFR2, thereby preventing the binding of its ligand VEGF. This may result in the inhibition of tumor angiogenesis and a decrease in tumor nutrient supply. VEGFR2 is a pro-angiogenic growth factor receptor -

Emerging Role of Tumor Cell Plasticity in Modifying Therapeutic Response
Signal Transduction and Targeted Therapy www.nature.com/sigtrans REVIEW ARTICLE OPEN Emerging role of tumor cell plasticity in modifying therapeutic response Siyuan Qin1, Jingwen Jiang1,YiLu 2,3, Edouard C. Nice4, Canhua Huang1,5, Jian Zhang2,3 and Weifeng He6,7 Resistance to cancer therapy is a major barrier to cancer management. Conventional views have proposed that acquisition of resistance may result from genetic mutations. However, accumulating evidence implicates a key role of non-mutational resistance mechanisms underlying drug tolerance, the latter of which is the focus that will be discussed here. Such non-mutational processes are largely driven by tumor cell plasticity, which renders tumor cells insusceptible to the drug-targeted pathway, thereby facilitating the tumor cell survival and growth. The concept of tumor cell plasticity highlights the significance of re-activation of developmental programs that are closely correlated with epithelial–mesenchymal transition, acquisition properties of cancer stem cells, and trans- differentiation potential during drug exposure. From observations in various cancers, this concept provides an opportunity for investigating the nature of anticancer drug resistance. Over the years, our understanding of the emerging role of phenotype switching in modifying therapeutic response has considerably increased. This expanded knowledge of tumor cell plasticity contributes to developing novel therapeutic strategies or combination therapy regimens using available anticancer drugs, which are likely to -
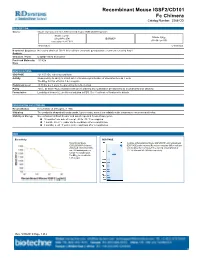
Recombinant Mouse IGSF2/CD101 Fc Chimera Catalog Number: 3368-CD
Recombinant Mouse IGSF2/CD101 Fc Chimera Catalog Number: 3368-CD DESCRIPTION Source Mouse myeloma cell line, NS0-derived mouse IGSF2/CD101 protein Mouse CD101 Mouse IgG (Gln21-Phe974) IEGRMDP 2a (Glu98-Lys330) Accession # A8E0Y8 N-terminus C-terminus N-terminal Sequence No results obtained. Gln21 inferred from enzymatic pyroglutamate treatment revealing Arg22. Analysis Structure / Form Disulfide-linked homodimer Predicted Molecular 133 kDa Mass SPECIFICATIONS SDS-PAGE 121-150 kDa, reducing conditions Activity Measured by its ability to inhibit anti-CD3-induced proliferation of stimulated human T cells. The ED50 for this effect is 1.4-8.4 μg/mL. Endotoxin Level <0.10 EU per 1 μg of the protein by the LAL method. Purity >95%, by SDS-PAGE visualized with Silver Staining and quantitative densitometry by Coomassie® Blue Staining. Formulation Lyophilized from a 0.2 μm filtered solution in PBS. See Certificate of Analysis for details. PREPARATION AND STORAGE Reconstitution Reconstitute at 400 μg/mL in PBS. Shipping The product is shipped with polar packs. Upon receipt, store it immediately at the temperature recommended below. Stability & Storage Use a manual defrost freezer and avoid repeated freeze-thaw cycles. 12 months from date of receipt, -20 to -70 °C as supplied. 1 month, 2 to 8 °C under sterile conditions after reconstitution. 3 months, ≤ -20 °C under sterile conditions after reconstitution. DATA Bioactivity SDS-PAGE Recombinant Mouse 2 μg/lane of Recombinant Mouse IGSF2/CD101 was resolved with IGSF2/CD101 Fc Chimera SDS-PAGE under reducing (R) and non-reducing (NR) conditions (Catalog # 3368-CD) inhibits and visualized by Coomassie® Blue staining, showing bands at anti-CD3 antibody induced 121-150 kDa and 240-280 kDa, respectively. -

2017 Immuno-Oncology Medicines in Development
2017 Immuno-Oncology Medicines in Development Adoptive Cell Therapies Drug Name Organization Indication Development Phase ACTR087 + rituximab Unum Therapeutics B-cell lymphoma Phase I (antibody-coupled T-cell receptor Cambridge, MA www.unumrx.com immunotherapy + rituximab) AFP TCR Adaptimmune liver Phase I (T-cell receptor cell therapy) Philadelphia, PA www.adaptimmune.com anti-BCMA CAR-T cell therapy Juno Therapeutics multiple myeloma Phase I Seattle, WA www.junotherapeutics.com Memorial Sloan Kettering New York, NY anti-CD19 "armored" CAR-T Juno Therapeutics recurrent/relapsed chronic Phase I cell therapy Seattle, WA lymphocytic leukemia (CLL) www.junotherapeutics.com Memorial Sloan Kettering New York, NY anti-CD19 CAR-T cell therapy Intrexon B-cell malignancies Phase I Germantown, MD www.dna.com ZIOPHARM Oncology www.ziopharm.com Boston, MA anti-CD19 CAR-T cell therapy Kite Pharma hematological malignancies Phase I (second generation) Santa Monica, CA www.kitepharma.com National Cancer Institute Bethesda, MD Medicines in Development: Immuno-Oncology 1 Adoptive Cell Therapies Drug Name Organization Indication Development Phase anti-CEA CAR-T therapy Sorrento Therapeutics liver metastases Phase I San Diego, CA www.sorrentotherapeutics.com TNK Therapeutics San Diego, CA anti-PSMA CAR-T cell therapy TNK Therapeutics cancer Phase I San Diego, CA www.sorrentotherapeutics.com Sorrento Therapeutics San Diego, CA ATA520 Atara Biotherapeutics multiple myeloma, Phase I (WT1-specific T lymphocyte South San Francisco, CA plasma cell leukemia www.atarabio.com -

Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association Between BMI and Adult-Onset Non- Atopic
Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association between BMI and Adult-Onset Non- Atopic Asthma Ayoung Jeong 1,2, Medea Imboden 1,2, Akram Ghantous 3, Alexei Novoloaca 3, Anne-Elie Carsin 4,5,6, Manolis Kogevinas 4,5,6, Christian Schindler 1,2, Gianfranco Lovison 7, Zdenko Herceg 3, Cyrille Cuenin 3, Roel Vermeulen 8, Deborah Jarvis 9, André F. S. Amaral 9, Florian Kronenberg 10, Paolo Vineis 11,12 and Nicole Probst-Hensch 1,2,* 1 Swiss Tropical and Public Health Institute, 4051 Basel, Switzerland; [email protected] (A.J.); [email protected] (M.I.); [email protected] (C.S.) 2 Department of Public Health, University of Basel, 4001 Basel, Switzerland 3 International Agency for Research on Cancer, 69372 Lyon, France; [email protected] (A.G.); [email protected] (A.N.); [email protected] (Z.H.); [email protected] (C.C.) 4 ISGlobal, Barcelona Institute for Global Health, 08003 Barcelona, Spain; [email protected] (A.-E.C.); [email protected] (M.K.) 5 Universitat Pompeu Fabra (UPF), 08002 Barcelona, Spain 6 CIBER Epidemiología y Salud Pública (CIBERESP), 08005 Barcelona, Spain 7 Department of Economics, Business and Statistics, University of Palermo, 90128 Palermo, Italy; [email protected] 8 Environmental Epidemiology Division, Utrecht University, Institute for Risk Assessment Sciences, 3584CM Utrecht, Netherlands; [email protected] 9 Population Health and Occupational Disease, National Heart and Lung Institute, Imperial College, SW3 6LR London, UK; [email protected] (D.J.); [email protected] (A.F.S.A.) 10 Division of Genetic Epidemiology, Medical University of Innsbruck, 6020 Innsbruck, Austria; [email protected] 11 MRC-PHE Centre for Environment and Health, School of Public Health, Imperial College London, W2 1PG London, UK; [email protected] 12 Italian Institute for Genomic Medicine (IIGM), 10126 Turin, Italy * Correspondence: [email protected]; Tel.: +41-61-284-8378 Int. -

What Lessons Can We Learn from 20 Years of Chemokine T D Di ? Receptor
What lessons can we learn from 20 years of chemokine receptdtor drug discovery? John G. Cumming, PhD 5th RSC / SCI symposium on GPCRs in Medicinal Chemistry 15th-17th September 2014, Actelion, Allschwil, Basel, Switzerland Outline Background: chemokines and their receptors Chemokine receptor drug discovery and development Emerging opportunities for chemokine drug discovery Conclusions and learning Chemokines and chemokine receptors CXC(α) • Chemokines (chemoattractant cytokines) are 70-120 aa proteins • 44 chemokines in 4 major families and 22 chemokine receptors in human genome • ‘Cell positioning system’ in the body • Many receptors bind multiple ligands • Many ligands bind multiple receptors Chemotaxis Human monocytes + CCL2 (red) Volpe et al. PLoS ONE 2012, 7(5), e37208 CCR2 antagonists inhibit chemotaxis and infiltration Vasculature CCL2 release Spinal or Peripheral Tissue Recruited monocyte Site of CCL2 release CCR2 antagonists inhibit chemotaxis and infiltration CCR2 antagonist Circulating monocyte CCL2 release CCL2 release from peripheral injury site or central PAF terminals Role of chemokine system in pathophysiology • Potential role in inflammatory and autoimmune diseases: Multiple sclerosis, Rheumatoid arthritis, COPD, allergic asthma, IBD, psoriasis - Expression levels of chemokines and receptors in relevant tissues and organs of patients and animal disease models - Mouse knockout ppyphenotype in disease models • Established role in HIV infection Katschke et al., 2001 Arthritis Rheum, 44, 1022 - CCR5 and CXCR4 act as HIV-1