Chronic Myeloid Leukemia: Mechanisms of Blastic Transformation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse
Welcome to More Choice CD Marker Handbook For more information, please visit: Human bdbiosciences.com/eu/go/humancdmarkers Mouse bdbiosciences.com/eu/go/mousecdmarkers Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse CD3 CD3 CD (cluster of differentiation) molecules are cell surface markers T Cell CD4 CD4 useful for the identification and characterization of leukocytes. The CD CD8 CD8 nomenclature was developed and is maintained through the HLDA (Human Leukocyte Differentiation Antigens) workshop started in 1982. CD45R/B220 CD19 CD19 The goal is to provide standardization of monoclonal antibodies to B Cell CD20 CD22 (B cell activation marker) human antigens across laboratories. To characterize or “workshop” the antibodies, multiple laboratories carry out blind analyses of antibodies. These results independently validate antibody specificity. CD11c CD11c Dendritic Cell CD123 CD123 While the CD nomenclature has been developed for use with human antigens, it is applied to corresponding mouse antigens as well as antigens from other species. However, the mouse and other species NK Cell CD56 CD335 (NKp46) antibodies are not tested by HLDA. Human CD markers were reviewed by the HLDA. New CD markers Stem Cell/ CD34 CD34 were established at the HLDA9 meeting held in Barcelona in 2010. For Precursor hematopoetic stem cell only hematopoetic stem cell only additional information and CD markers please visit www.hcdm.org. Macrophage/ CD14 CD11b/ Mac-1 Monocyte CD33 Ly-71 (F4/80) CD66b Granulocyte CD66b Gr-1/Ly6G Ly6C CD41 CD41 CD61 (Integrin b3) CD61 Platelet CD9 CD62 CD62P (activated platelets) CD235a CD235a Erythrocyte Ter-119 CD146 MECA-32 CD106 CD146 Endothelial Cell CD31 CD62E (activated endothelial cells) Epithelial Cell CD236 CD326 (EPCAM1) For Research Use Only. -

Targeting Non-Oncogene Addiction for Cancer Therapy
biomolecules Review Targeting Non-Oncogene Addiction for Cancer Therapy Hae Ryung Chang 1,*,†, Eunyoung Jung 1,†, Soobin Cho 1, Young-Jun Jeon 2 and Yonghwan Kim 1,* 1 Department of Biological Sciences and Research Institute of Women’s Health, Sookmyung Women’s University, Seoul 04310, Korea; [email protected] (E.J.); [email protected] (S.C.) 2 Department of Integrative Biotechnology, Sungkyunkwan University, Suwon 16419, Korea; [email protected] * Correspondence: [email protected] (H.R.C.); [email protected] (Y.K.); Tel.: +82-2-710-9552 (H.R.C.); +82-2-710-9552 (Y.K.) † These authors contributed equally. Abstract: While Next-Generation Sequencing (NGS) and technological advances have been useful in identifying genetic profiles of tumorigenesis, novel target proteins and various clinical biomarkers, cancer continues to be a major global health threat. DNA replication, DNA damage response (DDR) and repair, and cell cycle regulation continue to be essential systems in targeted cancer therapies. Although many genes involved in DDR are known to be tumor suppressor genes, cancer cells are often dependent and addicted to these genes, making them excellent therapeutic targets. In this review, genes implicated in DNA replication, DDR, DNA repair, cell cycle regulation are discussed with reference to peptide or small molecule inhibitors which may prove therapeutic in cancer patients. Additionally, the potential of utilizing novel synthetic lethal genes in these pathways is examined, providing possible new targets for future therapeutics. Specifically, we evaluate the potential of TONSL as a novel gene for targeted therapy. Although it is a scaffold protein with no known enzymatic activity, the strategy used for developing PCNA inhibitors can also be utilized to target TONSL. -
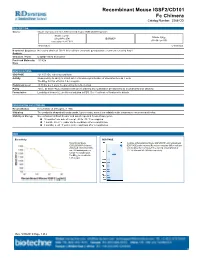
Recombinant Mouse IGSF2/CD101 Fc Chimera Catalog Number: 3368-CD
Recombinant Mouse IGSF2/CD101 Fc Chimera Catalog Number: 3368-CD DESCRIPTION Source Mouse myeloma cell line, NS0-derived mouse IGSF2/CD101 protein Mouse CD101 Mouse IgG (Gln21-Phe974) IEGRMDP 2a (Glu98-Lys330) Accession # A8E0Y8 N-terminus C-terminus N-terminal Sequence No results obtained. Gln21 inferred from enzymatic pyroglutamate treatment revealing Arg22. Analysis Structure / Form Disulfide-linked homodimer Predicted Molecular 133 kDa Mass SPECIFICATIONS SDS-PAGE 121-150 kDa, reducing conditions Activity Measured by its ability to inhibit anti-CD3-induced proliferation of stimulated human T cells. The ED50 for this effect is 1.4-8.4 μg/mL. Endotoxin Level <0.10 EU per 1 μg of the protein by the LAL method. Purity >95%, by SDS-PAGE visualized with Silver Staining and quantitative densitometry by Coomassie® Blue Staining. Formulation Lyophilized from a 0.2 μm filtered solution in PBS. See Certificate of Analysis for details. PREPARATION AND STORAGE Reconstitution Reconstitute at 400 μg/mL in PBS. Shipping The product is shipped with polar packs. Upon receipt, store it immediately at the temperature recommended below. Stability & Storage Use a manual defrost freezer and avoid repeated freeze-thaw cycles. 12 months from date of receipt, -20 to -70 °C as supplied. 1 month, 2 to 8 °C under sterile conditions after reconstitution. 3 months, ≤ -20 °C under sterile conditions after reconstitution. DATA Bioactivity SDS-PAGE Recombinant Mouse 2 μg/lane of Recombinant Mouse IGSF2/CD101 was resolved with IGSF2/CD101 Fc Chimera SDS-PAGE under reducing (R) and non-reducing (NR) conditions (Catalog # 3368-CD) inhibits and visualized by Coomassie® Blue staining, showing bands at anti-CD3 antibody induced 121-150 kDa and 240-280 kDa, respectively. -

1078-0432.CCR-20-1706.Full.Pdf
Author Manuscript Published OnlineFirst on June 29, 2020; DOI: 10.1158/1078-0432.CCR-20-1706 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Gastrointestinal stromal tumor: challenges and opportunities for a new decade César Serrano1,2, Suzanne George3 1Sarcoma Translational Research Laboratory, Vall d’Hebron Institute of Oncology, Barcelona, Spain. 2Department of Medical Oncology, Vall d’Hebron University Hospital, Barcelona, Spain. 3Department of Medical Oncology, Sarcoma Center, Dana-Farber Cancer Institute, Boston, US. Running title: Review on gastrointestinal stromal tumor Key words: Avapritinib; circulating tumor DNA; gastrointestinal stromal tumor; imatinib; regorafenib; ripretinib; sunitinib. Financial support: This work was supported by grants (to C.S.) from SARC Career Development Award, FIS ISCIII (PI19/01271), PERIS 2018 (SLT006/17/221) and FERO Foundation. Conflicts of interest: C.S. has received research grants from Deciphera Pharmaceuticals, Bayer AG and Pfizer, Inc; consulting fees (advisory role) from Deciphera Pharmaceuticals and Blueprint 1 Downloaded from clincancerres.aacrjournals.org on October 1, 2021. © 2020 American Association for Cancer Research. Author Manuscript Published OnlineFirst on June 29, 2020; DOI: 10.1158/1078-0432.CCR-20-1706 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Medicines; payment for lectures from Bayer AG and Blueprint Medicines; and travel grants from Pharmamar, Pfizer, Bayer AG, Novartis and Lilly. S.G. has received research funding to her institution from Blueprint Medicines, Deciphera Pharmaceutical, Bayer AG, Pfizer, Novartis; consulting fees (advisory role) from Blueprint Medicines, Deciphera Pharmaceuticals, Eli Lilly, Bayer AG. Corresponding author: Dr. César Serrano, Vall d’Hebron Institute of Oncology (VHIO), Vall d’Hebron University Hospital. -

Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association Between BMI and Adult-Onset Non- Atopic
Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association between BMI and Adult-Onset Non- Atopic Asthma Ayoung Jeong 1,2, Medea Imboden 1,2, Akram Ghantous 3, Alexei Novoloaca 3, Anne-Elie Carsin 4,5,6, Manolis Kogevinas 4,5,6, Christian Schindler 1,2, Gianfranco Lovison 7, Zdenko Herceg 3, Cyrille Cuenin 3, Roel Vermeulen 8, Deborah Jarvis 9, André F. S. Amaral 9, Florian Kronenberg 10, Paolo Vineis 11,12 and Nicole Probst-Hensch 1,2,* 1 Swiss Tropical and Public Health Institute, 4051 Basel, Switzerland; [email protected] (A.J.); [email protected] (M.I.); [email protected] (C.S.) 2 Department of Public Health, University of Basel, 4001 Basel, Switzerland 3 International Agency for Research on Cancer, 69372 Lyon, France; [email protected] (A.G.); [email protected] (A.N.); [email protected] (Z.H.); [email protected] (C.C.) 4 ISGlobal, Barcelona Institute for Global Health, 08003 Barcelona, Spain; [email protected] (A.-E.C.); [email protected] (M.K.) 5 Universitat Pompeu Fabra (UPF), 08002 Barcelona, Spain 6 CIBER Epidemiología y Salud Pública (CIBERESP), 08005 Barcelona, Spain 7 Department of Economics, Business and Statistics, University of Palermo, 90128 Palermo, Italy; [email protected] 8 Environmental Epidemiology Division, Utrecht University, Institute for Risk Assessment Sciences, 3584CM Utrecht, Netherlands; [email protected] 9 Population Health and Occupational Disease, National Heart and Lung Institute, Imperial College, SW3 6LR London, UK; [email protected] (D.J.); [email protected] (A.F.S.A.) 10 Division of Genetic Epidemiology, Medical University of Innsbruck, 6020 Innsbruck, Austria; [email protected] 11 MRC-PHE Centre for Environment and Health, School of Public Health, Imperial College London, W2 1PG London, UK; [email protected] 12 Italian Institute for Genomic Medicine (IIGM), 10126 Turin, Italy * Correspondence: [email protected]; Tel.: +41-61-284-8378 Int. -

Anti-Aging: Senolytics Or Gerostatics (Unconventional View)
www.oncotarget.com Oncotarget, 2021, Vol. 12, (No. 18), pp: 1821-1835 Review Anti-aging: senolytics or gerostatics (unconventional view) Mikhail V. Blagosklonny1 1Roswell Park Cancer Institute, Buffalo, NY 14263, USA Correspondence to: Mikhail V. Blagosklonny, email: [email protected], [email protected] Keywords: geroscience; senolytics; hyperfunction theory; aging; sirolimus Received: June 07, 2021 Accepted: July 05, 2021 Published: August 31, 2021 Copyright: © 2021 Blagosklonny. This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Senolytics are basically anti-cancer drugs, repurposed to kill senescent cells selectively. It is even more difficult to selectively kill senescent cells than to kill cancer cells. Based on lessons of cancer therapy, here I suggest how to exploit oncogene-addiction and to combine drugs to achieve selectivity. However, even if selective senolytic combinations will be developed, there is little evidence that a few senescent cells are responsible for organismal aging. I also discuss gerostatics, such as rapamycin and other rapalogs, pan-mTOR inhibitors, dual PI3K/mTOR inhibitors, which inhibit growth- and aging-promoting pathways. Unlike senolytics, gerostatics do not kill cells but slow down cellular geroconversion to senescence. Numerous studies demonstrated that inhibition of the mTOR pathways by any means (genetic, pharmacological and dietary) extends lifespan. Currently, only two studies demonstrated that senolytics (fisetin and a combination Dasatinib plus Quercetin) extend lifespan in mice. These senolytics slightly inhibit the mTOR pathway. Thus, life extension by these senolytics can be explained by their slight rapamycin-like (gerostatic) effects. -
Oncogene Addiction As a Rationale for Targeted Anti-Cancer Therapy in Hepatocellular Carcinoma
호암상 수상 기념특강 Oncogene addiction as a rationale for targeted anti-cancer therapy in hepatocellular carcinoma Dae-Ghon Kim Division of Gastroenterology and Hepatology, Department of Internal Medicine, Chonbuk National University Medical School and Hospital, Jeonju, Jeonbuk, Korea Abstract The concept of oncogene addiction was first introduced by Bernard Weinstein in 2000, with particular reference to the observation that some cyclin D-overexpressing cancers reverse their malignant phenotype upon cyclin-D deple- tion by means of RNA interference. It postulates that some tumours rely on one single dominant oncogene for growth and survival, so that inhibition of this specific oncogene is sufficient to halt the neoplastic phenotype. A large amount of evidence has proven the pervasive power of this notion, both in basic research and in therapeutic applications. Application of this concept to the clinical setting has achieved variable success in some various cancer types, including chronic myelogeneous leukaemia harbouring the BCR-ABL translocation, Erb2 overexpressing breast cancer, and non-small cell lung cancer harbouring a subset of EGFR mutations (Table1). However, in the face of such a considerable body of knowledge, the intimate molecular mechanisms mediating this phenomenon remain elusive. At the clinical level, successful translation of the oncogene addiction model into the rational and effective design of targeted therapeutics against individual oncoproteins still faces major obstacles, mainly due to the emergence of escape mechanisms and drug resistance. Sorafenib, a tyrosine kinase inhibitor (TKI), has demon- strated clinical efficacy in patients with HCC. Studies in patients with lung, breast, or colorectal cancers indicated that the genetic heterogeneity of cancer cells within a tumor affect its response to therapeutics designed to target specific molecules. -

Datasheet: MCA2236F Product Details
Datasheet: MCA2236F Description: MOUSE ANTI HUMAN CD101:FITC Specificity: CD101 Format: FITC Product Type: Monoclonal Antibody Clone: BB27 Isotype: IgG1 Quantity: 0.1 mg Product Details Applications This product has been reported to work in the following applications. This information is derived from testing within our laboratories, peer-reviewed publications or personal communications from the originators. Please refer to references indicated for further information. For general protocol recommendations, please visit www.bio-rad-antibodies.com/protocols. Yes No Not Determined Suggested Dilution Flow Cytometry 1/5 - 1/10 Where this antibody has not been tested for use in a particular technique this does not necessarily exclude its use in such procedures. Suggested working dilutions are given as a guide only. It is recommended that the user titrates the antibody for use their own system using appropriate negative/positive controls. Target Species Human Product Form Purified IgG conjugated to Fluorescein Isothiocyanate Isomer 1 (FITC) - liquid Max Ex/Em Fluorophore Excitation Max (nm) Emission Max (nm) FITC 490 525 Preparation Purified IgG prepared by affinity chromatography on Protein G from tissue culture supernatant Buffer Solution Phosphate buffered saline Preservative 0.09% Sodium Azide Stabilisers 1% Bovine Serum Albumin Approx. Protein IgG concentration 0.1 mg/ml Concentrations Immunogen Human thymic clone B12. External Database Links UniProt: Q93033 Related reagents Entrez Gene: Page 1 of 3 9398 CD101 Related reagents Synonyms EWI101, IGSF2, V7 Specificity Mouse anti Human CD101 antibody, clone BB27 recognizes human CD101, also known as Immunoglobulin superfamily member 2 (IgSF2), . Cell surface glycoprotein V7, Glu-Trp-Ile EWI motif-containing protein 101 or EWI-101. -

Emerging Insights of Tumor Heterogeneity and Drug Resistance Mechanisms in Lung Cancer Targeted Therapy Zuan-Fu Lim1,2,3 and Patrick C
Lim and Ma Journal of Hematology & Oncology (2019) 12:134 https://doi.org/10.1186/s13045-019-0818-2 REVIEW Open Access Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy Zuan-Fu Lim1,2,3 and Patrick C. Ma3* Abstract The biggest hurdle to targeted cancer therapy is the inevitable emergence of drug resistance. Tumor cells employ different mechanisms to resist the targeting agent. Most commonly in EGFR-mutant non-small cell lung cancer, secondary resistance mutations on the target kinase domain emerge to diminish the binding affinity of first- and second-generation inhibitors. Other alternative resistance mechanisms include activating complementary bypass pathways and phenotypic transformation. Sequential monotherapies promise to temporarily address the problem of acquired drug resistance, but evidently are limited by the tumor cells’ ability to adapt and evolve new resistance mechanisms to persist in the drug environment. Recent studies have nominated a model of drug resistance and tumor progression under targeted therapy as a result of a small subpopulation of cells being able to endure the drug (minimal residual disease cells) and eventually develop further mutations that allow them to regrow and become the dominant population in the therapy-resistant tumor. This subpopulation of cells appears to have developed through a subclonal event, resulting in driver mutations different from the driver mutation that is tumor- initiating in the most common ancestor. As such, an understanding of intratumoral heterogeneity—the driving force behind minimal residual disease—is vital for the identification of resistance drivers that results from branching evolution. Currently available methods allow for a more comprehensive and holistic analysis of tumor heterogeneity in that issues associated with spatial and temporal heterogeneity can now be properly addressed. -

Discovery of Dihydroartemisinin and Dasatinib Drug Combination To
University of Tennessee Health Science Center UTHSC Digital Commons Theses and Dissertations (ETD) College of Graduate Health Sciences 8-2012 Discovery of Dihydroartemisinin and Dasatinib Drug Combination to Cure Pooroutcome BCR- ABL+ Acute Lymphoblastic Leukemia Harpreet Singh University of Tennessee Health Science Center Follow this and additional works at: https://dc.uthsc.edu/dissertations Part of the Medical Sciences Commons, Neoplasms Commons, Pharmaceutical Preparations Commons, and the Pharmaceutics and Drug Design Commons Recommended Citation Singh, Harpreet , "Discovery of Dihydroartemisinin and Dasatinib Drug Combination to Cure Pooroutcome BCR-ABL+ Acute Lymphoblastic Leukemia" (2012). Theses and Dissertations (ETD). Paper 249. http://dx.doi.org/10.21007/etd.cghs.2012.0292. This Dissertation is brought to you for free and open access by the College of Graduate Health Sciences at UTHSC Digital Commons. It has been accepted for inclusion in Theses and Dissertations (ETD) by an authorized administrator of UTHSC Digital Commons. For more information, please contact [email protected]. Discovery of Dihydroartemisinin and Dasatinib Drug Combination to Cure Pooroutcome BCR-ABL+ Acute Lymphoblastic Leukemia Document Type Dissertation Degree Name Doctor of Philosophy (PhD) Program Biomedical Sciences Track Cancer and Developmental Biology Research Advisor Gerard P. Zambetti, Ph.D. (for Richard T. Williams, M.D., Ph.D.) Committee Suzanne J. Baker, Ph.D. R. Kiplin Guy, Ph.D. Sima Jeha, Ph.D. Rajendra S. Raghow, Ph.D. DOI 10.21007/etd.cghs.2012.0292 This dissertation is available at UTHSC Digital Commons: https://dc.uthsc.edu/dissertations/249 Discovery of Dihydroartemisinin and Dasatinib Drug Combination to Cure Poor-outcome BCR-ABL+ Acute Lymphoblastic Leukemia A Dissertation Presented for The Graduate Studies Council The University of Tennessee Health Science Center In Partial Fulfillment Of the Requirements for the Degree Doctor of Philosophy From The University of Tennessee By Harpreet Singh August 2012 Copyright © 2012 by Harpreet Singh. -

Human CD Marker Chart Reviewed by HLDA1 Bdbiosciences.Com/Cdmarkers
BD Biosciences Human CD Marker Chart Reviewed by HLDA1 bdbiosciences.com/cdmarkers 23-12399-01 CD Alternative Name Ligands & Associated Molecules T Cell B Cell Dendritic Cell NK Cell Stem Cell/Precursor Macrophage/Monocyte Granulocyte Platelet Erythrocyte Endothelial Cell Epithelial Cell CD Alternative Name Ligands & Associated Molecules T Cell B Cell Dendritic Cell NK Cell Stem Cell/Precursor Macrophage/Monocyte Granulocyte Platelet Erythrocyte Endothelial Cell Epithelial Cell CD Alternative Name Ligands & Associated Molecules T Cell B Cell Dendritic Cell NK Cell Stem Cell/Precursor Macrophage/Monocyte Granulocyte Platelet Erythrocyte Endothelial Cell Epithelial Cell CD1a R4, T6, Leu6, HTA1 b-2-Microglobulin, CD74 + + + – + – – – CD93 C1QR1,C1qRP, MXRA4, C1qR(P), Dj737e23.1, GR11 – – – – – + + – – + – CD220 Insulin receptor (INSR), IR Insulin, IGF-2 + + + + + + + + + Insulin-like growth factor 1 receptor (IGF1R), IGF-1R, type I IGF receptor (IGF-IR), CD1b R1, T6m Leu6 b-2-Microglobulin + + + – + – – – CD94 KLRD1, Kp43 HLA class I, NKG2-A, p39 + – + – – – – – – CD221 Insulin-like growth factor 1 (IGF-I), IGF-II, Insulin JTK13 + + + + + + + + + CD1c M241, R7, T6, Leu6, BDCA1 b-2-Microglobulin + + + – + – – – CD178, FASLG, APO-1, FAS, TNFRSF6, CD95L, APT1LG1, APT1, FAS1, FASTM, CD95 CD178 (Fas ligand) + + + + + – – IGF-II, TGF-b latency-associated peptide (LAP), Proliferin, Prorenin, Plasminogen, ALPS1A, TNFSF6, FASL Cation-independent mannose-6-phosphate receptor (M6P-R, CIM6PR, CIMPR, CI- CD1d R3G1, R3 b-2-Microglobulin, MHC II CD222 Leukemia -

Oncogene Addiction As a Foundational Rationale for Targeted Anti-Cancer Therapy: Promises and Perils
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Institutional Research Information System University of Turin Review Oncogene addiction and targeted anti-cancer therapy Oncogene addiction as a foundational rationale for targeted anti-cancer therapy: promises and perils Davide Torti1, Livio Trusolino1* Keywords: DNA damage; drug development; oncogene addiction; targeted therapies; tyrosine kinases DOI 10.1002/emmm.201100176 Received April 15, 2011 / Revised July 07, 2011 / Accepted August 04, 2011 A decade has elapsed since the concept of oncogene addiction was first proposed. It postulates that – despite the diverse array of genetic lesions typical of cancer – some tumours rely on one single dominant oncogene for growth and survival, so that Introduction inhibition of this specific oncogene is sufficient to halt the neoplastic phenotype. A large amount of evidence has proven the pervasive power of this notion, both in Coordinated efforts by the Interna- tional Cancer Genome Consortium basic research and in therapeutic applications. However, in the face of such a and other Institutions have started to considerable body of knowledge, the intimate molecular mechanisms mediating this reveal the complex and heterogeneous phenomenon remain elusive. At the clinical level, successful translation of the ‘genomic landscapes’ of many human oncogene addiction model into the rational and effective design of targeted cancer types with unprecedented accu- therapeutics against individual oncoproteins still faces major obstacles, mainly racy (Hudson et al, 2010; Stratton et al, due to the emergence of escape mechanisms and drug resistance. Here, we offer 2009). A variegate repertoire of genetic an overview of the relevant literature, encompassing both biological aspects and aberrations – including point muta- recent clinical insights.