Liposomal Targeting of Antimicrobial Agents to Bacterial Infections by Raymond Michel Schiffelers
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

2008 Annual Report Committee: Keith A
TOWN OF NORTH ATTLEBOROUGH 2008 ANNUAL REPORT TOWN OF NORTH ATTLEBOROUGH 2008 ANNUAL REPORT Cover Photography – Patty Hitchcock: “The Gasholder” Winning Photograph in the Massachusetts Municipal Association’s 2009 State Calendar Contest 2008 Annual Report Committee: Keith A. Mueller and Judith Chafetz-Sulfaro Printing: Sir Speedy Printing, North Attleborough, MA Editor and Coordinator: Judith Chafetz-Sulfaro Photographs courtesy of Patty Hitchcock The 2008 Annual Report of the Town of North Attleborough has been dedicated to the memory of the employees, retirees, and committee members who passed away in 2008. Your contributions to our town have been immeasurable. You remain in our thoughts and will live on in our hearts forever. Rest in Peace, Dear Friends. Town of North Attleborough Employees, Retirees, and Committee Members Who Passed Away in 2008 DEPARTMENT, BOARD NAME DATE OR COMMITTEE Edward G. Lambert, Jr. January 10, 2008 Chairman, Retirement Board George E. Landry January 28, 2008 Highway Department; RTM Carol Paquette February 15, 2008 Town Clerk/Tax Collector Renaldo J. Chelotti February 23, 2008 School Department Harriet Wilmarth February 29, 2008 School Department John Flynn March 2, 2008 Bus Driver, Council on Aging Arthur Gelichauf, Jr. March 6, 2008 Town Accountant William J. McKeon March 29, 2008 Fire Department Gerald F. Dugan May 12, 2008 NAED Susan Nelson May 21, 2008 Board of Selectmen, 1st female Gilberte M. Hebert September 7, 2008 Secretary, Retirement Board John L. Thorp September 7, 2008 Fire Commissioner; RTM Town Moderator, Advisory Board to Veterans’ Council Katherine E. Dupelle October 24, 2008 School Department Lester E. Caldwell November 24, 2008 Chief, North Attleborough Fire Department; Board of Selectmen Eugene R. -

Récidive À Long Terme Du Syndrome De Vasoconstriction Cérébral Réversible : Suivi Prospectif De 173 Patients Rosalie Boitet
Récidive à long terme du syndrome de vasoconstriction cérébral réversible : suivi prospectif de 173 patients Rosalie Boitet To cite this version: Rosalie Boitet. Récidive à long terme du syndrome de vasoconstriction cérébral réversible : suivi prospectif de 173 patients. Médecine humaine et pathologie. 2018. dumas-02955583 HAL Id: dumas-02955583 https://dumas.ccsd.cnrs.fr/dumas-02955583 Submitted on 2 Oct 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution - NonCommercial - ShareAlike| 4.0 International License UNIVERSITÉ DE MONTPELLIER FACULTÉ DE MÉDECINE MONTPELLIER-NIMES THÈSE Pour obtenir le titre de DOCTEUR EN MÉDECINE Présentée et soutenue publiquement Par Rosalie BOITET Le 22 juin 2018 RÉCIDIVE A LONG TERME DU SYNDROME DE VASOCONSTRICTION CÉRÉBRAL RÉVERSIBLE : SUIVI PROSPECTIF DE 173 PATIENTS Directeur de thèse : Professeur Anne DUCROS JURY Président : Professeur Pierre LABAUGE Assesseurs : Professeur Anne DUCROS Professeur Éric THOUVENOT Docteur Caroline ARQUIZAN UNIVERSITÉ DE MONTPELLIER -

Download a Copy of the 264-Page Publication
2020 Department of Neurological Surgery Annual Report Reporting period July 1, 2019 through June 30, 2020 Table of Contents: Introduction .................................................................3 Faculty and Residents ...................................................5 Faculty ...................................................................6 Residents ...............................................................8 Stuart Rowe Lecturers .........................................10 Peter J. Jannetta Lecturers ................................... 11 Department Overview ............................................... 13 History ............................................................... 14 Goals/Mission .................................................... 16 Organization ...................................................... 16 Accomplishments of Note ................................ 29 Education Programs .................................................. 35 Faculty Biographies ................................................... 47 Resident Biographies ................................................171 Research ....................................................................213 Overview ...........................................................214 Investigator Research Summaries ................... 228 Research Grant Summary ................................ 242 Alumni: Past Residents ........................................... 249 Donations ................................................................ 259 Statistics -

Unclaimed Capital Credits 2017-19
DATE NAME 10/2/2017 FRANK ABATE 10/2/2017 CHRIS W ABEL 10/2/2017 EMIL ABEL 10/2/2017 ERIN STACY ABERNATHY 10/2/2017 CLEMENT ABILA 10/2/2017 CARRIE JEAN ABRAHAM 10/2/2017 CHERI LYNN ADAM 10/2/2017 MILTON S ADAMS 10/2/2017 SHIRLEY ADAMS 10/2/2017 VICKIE ADAMS 10/2/2017 CLYDE ADCOCK 10/2/2017 MARCOS S AIELLO 10/2/2017 LATEEF HASHIM AL-SARAJ I 10/2/2017 GORDON L ALBRIGHT ESTATE 10/2/2017 A T ALBRIGHT 10/2/2017 DEBBIE ALDERSON 10/2/2017 TINA ALEXANDER 10/2/2017 AUGUSTA ALFORD 10/2/2017 ANGELEAN JEAN ALLEN 10/2/2017 CHARLES H ALLEN 10/2/2017 JIM ALLISON ESTATE 10/2/2017 GARY PATRICK ALLISON 10/2/2017 NELDA F ALLRED 10/2/2017 RICHARD PAUL ALLS 10/2/2017 ALLTEL COMM INC 10/2/2017 JULIE B ALMQUIST 10/2/2017 GREGORY ALLEN ALTHOFF 10/2/2017 IRA M ALTMAN 10/2/2017 JUSTIN AMBURGEY 10/2/2017 STEPHEN L ANDERS 10/2/2017 G S ANDERSON SR 10/2/2017 ANGIE N ANDERSON 10/2/2017 BILLY GENE ANDERSON 10/2/2017 FRANK R ANDERSON 10/2/2017 J E ANDERSON 10/2/2017 JAMES O ANDERSON 10/2/2017 JERRY ANDERSON 10/2/2017 LELYA ANDERSON 10/2/2017 MARILYN J ANDERSON 10/2/2017 SHORTY ANDERSON 10/2/2017 MARGIE ANZ 10/2/2017 MICHAEL ARBUCKLE 10/2/2017 BRAZOS ARCOTTA 10/2/2017 LOUIS ARIAS JR 10/2/2017 ISMAEL ARIAS 10/2/2017 TERRY A ARMBRUSTER 10/2/2017 LESLIE T ARMSTRONG 10/2/2017 BILLY ARNDT 10/2/2017 BERTHA ARNOLD ESTATE 10/2/2017 WATSON C ARNOLD ESTATE 10/2/2017 DEBRA ARNOLD 10/2/2017 JESS ARNOLD 10/2/2017 MRS L J ARNOLD 10/2/2017 TINA R ARNOLD 10/2/2017 JAY BRANDON ARP 10/2/2017 TOMMY W ARP 10/2/2017 STEVEN C ASH 10/2/2017 MARGIE ASHBY 10/2/2017 SEABORN CHRISTOPHER ASHBY 10/2/2017 -

Elettorato Definitivo Del 07/07/2011<Br>Ricercatori
Ministero dell Istruzione,, dell ,Università e della Ricerca II sessione 2010 Elettorato Definitivo del 07/07/2011 Ricercatori 1 Elettorato Definitivo Settore: AGR/01 - Economia ed estimo rurale Bandi previsti:6 Numero di docenti necessari alla costituzione della lista dei sorteggiabili: 36 Elettorato attivo N° Cognome Nome Ateneo Facoltà Settore Nota ** Data di nascita Data di nomina 1 AMATA Francesco Univ. CATANIA AGRARIA AGR/01 30/07/1944 01/03/2006 2 ANTONELLI Gervasio Univ. URBINO ECONOMIA AGR/01 24/03/1946 01/11/1999 3 BANTERLE Alessandro Univ. MILANO AGRARIA AGR/01 STR. 05/07/1960 01/11/2010 4 BASILE Elisabetta ROMA "La Sapienza" ECONOMIA (*) AGR/01 01/08/1951 01/11/2001 5 BEGALLI Diego Univ. VERONA ECONOMIA AGR/01 23/09/1957 01/03/2000 6 BERNETTI Iacopo Univ. FIRENZE AGRARIA AGR/01 27/01/1963 01/11/2000 7 BERTAZZOLI Aldo Univ. BOLOGNA AGRARIA AGR/01 19/07/1959 01/11/2001 8 BOATTO Vasco Ladislao Univ. PADOVA AGRARIA AGR/01 27/06/1950 30/10/1986 9 BOVE Ettore Univ. BASILICATA ECONOMIA AGR/01 11/04/1947 01/11/1994 10 BRUNORI Gianluca Univ. PISA AGRARIA AGR/01 25/03/1960 30/12/2004 11 CANNATA Giovanni Univ. MOLISE ECONOMIA AGR/01 CUN 08/03/1947 01/11/1991 12 CARRA' Giuseppina Univ. CATANIA AGRARIA AGR/01 14/08/1951 01/11/1994 13 CASATI Dario Univ. MILANO AGRARIA AGR/01 18/09/1943 01/11/1980 14 CASINI Leonardo Univ. FIRENZE AGRARIA AGR/01 19/06/1958 01/11/1994 15 CECCHI Claudio ROMA "La Sapienza" ECONOMIA (*) AGR/01 29/09/1949 01/11/2000 16 CESARETTI Gian Paolo "Parthenope" NAPOLI ECONOMIA AGR/01 19/11/1943 22/04/1986 17 CHANG Ting Fa Margherita Univ. -

28776/2010 IC – Fls
1 COORDENADORES FCA Alvaro de Oliveira D´Antona FCM José Barreto Campello Carvalheira FE Dario Fiorentini FEA Helena Teixeira Godoy FEAGRI Zigomar Menezes de Souza FEEC Carlos Alberto de Castro Junior FEC Maria Cecília Amorim Teixeira da Silva FEF Antonio Carlos de Moraes FEM Kátia Lucchesi Cavalca Dedini FEQ Martin Aznar FOP Renata Cunha Matheus Rodrigues Garcia FT Regina Lúcia de Oliveira Moraes IA Emerson Luiz de Biaggi IB Helena Coutinho Franco de Oliveira IC Paulo Lício de Geus IE Fernando Sarti (Representando a CPG) IEL Fabio Akcelrud Durão IFCH Omar Ribeiro Thomaz IFGW Silvio Antonio Sachetto Vitiello IG Carlos Roberto de Souza Filho IMECC Luiz Koodi Hotta IQ Maria Isabel Felisberti REPRESENTAÇÃO DISCENTE – TITULARES E SUPLENTES FEM Ana Luisa Soubhia - Titular IQ Marcelo Fabiano André - Titular FEEC Hugo Valadares Siqueira - Titular FEM Eduardo Welzl - Suplente FEM Roberto Mac Intyer Simões – Suplente CONVIDADOS PERMANENTES DAC Antônio Faggiani PRP Ronaldo Aloise Pilli PREAC Mohamed Mostafa Habib ASSESSORA Rosana Aparecida Baeninger ASSESSORA Maria de Fátima Sonati ASSESSOR Munir Salomão Skaf 2 ORDEM DO DIA 1. REVALIDAÇÃO DE DIPLOMAS ESTRANGEIROS Para Aprovação a) PROC. Nº 01P-16550/2011 IC – MATTHIAS RUDOLF BRUST – “Docteur Parecer Favorável en Informatique” – Université du Luxembourg, Luxemburgo. Fls. 8 a 11 b) PROC. Nº 01P-17860/2011 IFGW – JAVIER FERNANDO RAMOS CARO Parecer Favorável – “Doctor en Ciencias Naturales (Física)” – La Universidad Industrial de Santander, Bucaramanga, Colombia. Fls. 12 a 15 c) PROC. Nº 01P-11000/2011 FEEC – DANIEL LAMBERTZ – “Docteur Parecer Favorável Spécialité Genie Biomedical” – Université de Technologie de Compiégne, França. Fls. 16 a 20 d) PROC. Nº 01P-27972/2010 FEEC – MEIRE CRISTINA FUGIHARA – Parecer Favorável “Doutorado em Engenharia Electrotécnica” – Universidade de Aveiro, Portugal. -
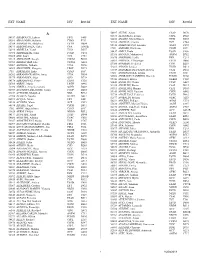
Ext Name Div Room Ext Name Div Room ______
EXT NAME DIV ROOM EXT NAME DIV ROOM __________________________________________________________________________________________________________________________ A 56407 ALUNG, Ashok CSAP D170 52673 ALVETRETI, Letizia CSPL D328 54157 ABABOUCH, Lahsen FIPX F405 54458 AMADO, Maria Blanca TCIB D503 55581 ABALSAMO, Stefania CIOO F713 53290 AMARAL, Cristina TCE C744 53264 ABBASSIAN, Abdolreza ESTD D804 55126 AMBROSIANO, Luciana AGDF C292 54511 ABBONDANZA, Carla CPA A143B 1709 AMEGEE, Emilienne CSSD SSC 52390 ABDELLA, Yesuf TCIA D557 56437 AMICI, Paolo ESSD C456 55396 ABDIRIZZAK, Tania FOED C474 53985 AMMATI, Mohammed AGPM B752 53485 ABE, Kaori TCE C751 54876 AMOROSO, Leslie ESN C214 54315 ABI NASSIF, Joseph CSDM B104 56891 AMROUK, El Mamoun ESTD D866 53963 ABI RACHED, Elie CPAM A281 53980 ANAMAN, Frederick CIOF B239 55087 ABITBOL, Nathalie TCIA D556 54213 ANAND, Sanjeev TCSD D611 56584 ABOU-RIZK, Margaret LEGP D371 53309 ANDARIAS DE PRADO, Rebeca CSAI D102 1716 ABRAHAM, Erika CSSD SSC 1725 ANDONOVSKA, Liljana CSSD SSC 56202 ABRAMO-GUARNA, Anna TCIA D554 53602 ANDRADE CIANFRINI, Graciela FOMD D468 55375 ABRAMOVA, Olga OSD D728 55742 ANELLO, Enrico OEKM C102 54178 ABRAMOVICI, Pierre CIOH C130 54441 ANGELINI, Chiara CPAP A487 56760 ABREU, Maria LEGD A442 54109 ANGELINI, Flavio CSAI D114 53296 ABRINA, Angelica Gavina OSPD B449 55313 ANGELONI, Marino CSAI D103 52709 ACCARDO-DELHOME, Jeanne FODP D435 53145 ANGELOZZI, Vanessa CSDU A042 53843 ACHOURI, Moujahed NRL B732 53720 ANGELUCCI, Federica ESTD D863 52294 ACOSTA, Natalia OEDD B452 52677 ANIBALDI, -

Editor: Maurizio Tonetti
ISSN 0303 – 6979 VOLUME 48, NUMBER 1, JANUARY 2021 Editor: Maurizio Tonetti Offi cial journal of the European Federa on of Periodontology Founded by the Bri sh, Dutch, French, German, Scandinavian and Swiss Socie es of Periodontology wileyonlinelibrary.com/journal/jcpe EDITOR-IN-CHIEF: EDITORIAL BOARD: P. Hujoel, Seattle, WA, USA D. W. Paquette, Chapel Hill, NC, USA Maurizio Tonetti P. Adriaens, Brussels, Belgium J. Hyman, Vienna, VA, USA G. Pini-Prato, Florence, Italy Journal of Clinical Periodontology J. Albandar, Philadelphia, PA, USA I. Ishikawa, Tokyo, Japan P. Preshaw, Newcastle upon Tyne, UK Editorial Office G. Armitage, San Francisco, CA, J. Jansen, Nijmegen, the Netherlands M. Ryder, San Francisco, CA, USA John Wiley & Sons Ltd USA P.-M. Jervøe-Storm, Bonn, Germany G. E. Salvi, Berne, Switzerland 9600 Garsington Road, Oxford D. Botticelli, Rimini, Italy L. J. Jin, Hong Kong SAR, China A. Schaefer, Kiel-Schleswig-Holstein, OX4 2DQ, UK P. Bouchard, Paris, France A. Kantarci, Boston, MA, USA Germany E-mail: [email protected] A. Braun, Marburg, Germany D. F. Kinane, Louisville, KY, USA D. A. Scott, Louisville, Kentucky, USA K. Buhlin, Huddinge, Sweden M. Kebschull, Bonn, Germany A. Sculean, Berne, Switzerland ASSOCIATE EDITORS: M. Christgau, Düsseldorf, Germany M. Klepp, Stavanger, Norway L. Shapira, Jerusalem, Israel T. Berglundh, Göteborg, Sweden P. Cortellini, Florence, Italy T. Kocher, Greifswald, Germany B. Stadlinger, Zürich, Switzerland I. Chapple, Birmingham, UK F. Cairo, Florence, Italy E. Lalla, New York, NY, USA A. Stavropoulos, Aarhus C, Denmark R. Demmer, New York, NY, USA G. Dahlen, Gothenburg, Sweden N. P. Lang, Berne, Switzerland D. Tatakis, Columbus, OH, USA G. -

Comparaison De Deux Séries À 10 Ans D'intervalle À Grenoble
Évolution de la traumatologie des sports d’hiver : comparaison de deux séries à 10 ans d’intervalle à Grenoble (1998-1999 et 2008-2009) Florent Vejux, Nicolas Picard To cite this version: Florent Vejux, Nicolas Picard. Évolution de la traumatologie des sports d’hiver : comparaison de deux séries à 10 ans d’intervalle à Grenoble (1998-1999 et 2008-2009). Médecine humaine et pathologie. 2012. dumas-00748586 HAL Id: dumas-00748586 https://dumas.ccsd.cnrs.fr/dumas-00748586 Submitted on 5 Nov 2012 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. AVERTISSEMENT Ce document est le fruit d'un long travail approuvé par le jury de soutenance et mis à disposition de l'ensemble de la communauté universitaire élargie. Il n’a pas été réévalué depuis la date de soutenance. Il est soumis à la propriété intellectuelle de l'auteur. Ceci implique une obligation de citation et de référencement lors de l’utilisation de ce document. D’autre part, toute contrefaçon, plagiat, reproduction illicite encourt une poursuite pénale. Contact au SICD1 de Grenoble : [email protected] LIENS LIENS Code de la Propriété Intellectuelle. articles L 122. -

E-Poster List
E-POSTER LIST The complete list of abstracts accepted as e-posters for the 29th EADV Congress, 29th – 31st October, 2020 Acne and Related Disorders, Hidradenitis Suppurativa P0001 Lupus Milliaris Disseminatus Faciei : A rare case treated with Isotretinoin Jesmin A. (DHAKA, Bangladesh) P0002 A comparative study of microneedling alone versus along with platelet rich plasma in acne scars Debbarman K., Gupta M. (Delhi, India) P0003 Serum levels of 25-hydroxyvitamin D and IL17 and their association with acne severity in patients with severe and very severe acne vulgaris Elkamshoushi A., Elneily D., Ismail S., Mahmoud H. (Alexandria, Egypt) P0006 The psychosocial impact of Hidradenitis suppurativa Vasileva S., Vasilev S., Vasilev B., Brishkoska Boshkovski V., Vasileva M. (Shtip, Macedonia) P0008 Myths, Perceptions and Practices in Acne: A Study on Adolescents and Young Adults Yorulmaz A., Yalçın B. (Ankara, Turkey) P0009 Adalimumab in severe hidradenitis suppurativa - A case report of a novel experiencr Balendran T., Gunasekera C. (Batticaloa, Sri Lanka) P0010 Relationship between acne vulgaris and insulin resistance markers and effect of isotretinoin on insulin resistance markers in patients with acne vulgaris: A systematic review and meta- analysis Raizada A. (Bhubaneswar , India) P0011 Systemic steroids in moderate-to-severe hidradenitis suppurativa Duarte B., Cunha N., Lencastre A., Cabete J. (Lisbon, Portugal) P0012 Ixekizumab restores clinical response in patients with secondary loss of efficacy from Adalimumab: results of a case series of 10 patients with Hidradenitis suppurativa Hilbring C., Augustin M., Girbig G., Kirsten N. (Hamburg, Germany) P0013 Microbiological assessment of macrolides and lincosamides effectiveness in the treatment of folliculitis and acne in Ukraine Voloshynovych M., Kutsyk R., Chmut V., Yurchyshyn O., Pavliuk N., Rusko H. -

41454 23-2 Legala
Government Gazette Staatskoerant REPUBLIC OF SOUTH AFRICA REPUBLIEK VAN SUID-AFRIKA February Vol. 632 Pretoria, 23 2018 Februarie No. 41454 PART 1 OF 2 LEGAL NOTICES A WETLIKE KENNISGEWINGS ISSN 1682-5843 N.B. The Government Printing Works will 41454 not be held responsible for the quality of “Hard Copies” or “Electronic Files” submitted for publication purposes 9 771682 584003 AIDS HELPLINE: 0800-0123-22 Prevention is the cure 2 No. 41454 GOVERNMENT GAZETTE, 23 FEBRUARY 2018 IMPORTANT NOTICE: THE GOVERNMENT PRINTING WORKS WILL NOT BE HELD RESPONSIBLE FOR ANY ERRORS THAT MIGHT OCCUR DUE TO THE SUBMISSION OF INCOMPLETE / INCORRECT / ILLEGIBLE COPY. NO FUTURE QUERIES WILL BE HANDLED IN CONNECTION WITH THE ABOVE. Table of Contents LEGAL NOTICES BUSINESS NOTICES • BESIGHEIDSKENNISGEWINGS Gauteng ....................................................................................................................................... 12 Eastern Cape / Oos-Kaap ................................................................................................................. 15 Free State / Vrystaat ........................................................................................................................ 15 KwaZulu-Natal ................................................................................................................................ 16 Limpopo ....................................................................................................................................... 17 Mpumalanga ................................................................................................................................. -
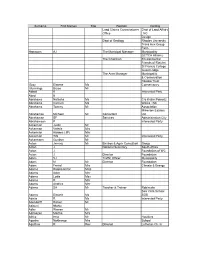
Nuclear-1 IAP Register RDEIR V2
Surname First Names Title Position Co/Org Land Claims Commissioner Dept of Land Affairs Office : NC Design Dept of Geology Rhodes University Trans Hex Group Farm Hanacom AJ The Municipal Manager Municipality GCTCA Alliance The Chairman Environmental Friends of Rietvlei St Francis College Guest Lodge The Area Manager Municipality & Conservation Rosalia Trust Bray Elmene Ms Conservancy Munnings Bruce Mr Abbott K Interested Party Abed N Abrahams Natasha Ms C/o Katrin Pobantz Abrahams Carmen Ms Works : NC Abrahams Tommy Mr Association Milnerton Estates Abrahamse Michael Mr Consultant Ltd Abrahamse SF Services Administration City Abrahamson P Interested Party Ackerman Justin Mr Ackerman Valerie Mrs Ackerman Hannes (JP) Mnr Ackerman WN Mr Interested Party Ackermann Gunther Mr Acton Jeremy Mr Environ & Agric Consultant Group Acton J National Secretary South Africa Acton J Foundation of WC Acton J Director Foundation Adam SJ Traffic Officer Municipality Adam M Mr Director Foundation Adam Ferrial Mrs Climate & Energy Adams Dideka Elizar Miss Adams Aden Mnr Adams Lydia Mev Adams R Mnr Adams Andries Mnr Adams SA Mr Teacher & Trainer Robinvale Sea Vista School Adams Elmarie Ms SGB Adatia R Ms Interested Party Adendorff Daniel Mr Adisuni Ntahlu Adler Warren Mr Adriaazen Martha Mrs Afrika Irma Mr Haulliers Agoshe Nokhanyo Mrs School Agullhas R Rev Director Lutheran Ch. In Ah Shene Carolyn Miss Advocacy Africa Airey D Interested Party Alba A Interested Party Albers Anthony Dr Projects / Nightsky Albertyn Janice Mrs Representative Botanical Society Alcock