The Relationship Between the Level of Anterior Cingulate Cortex
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Function of Cerebral Cortex
FUNCTION OF CEREBRAL CORTEX Course: Neuropsychology CC-6 (M.A PSYCHOLOGY SEM II); Unit I By Dr. Priyanka Kumari Assistant Professor Institute of Psychological Research and Service Patna University Contact No.7654991023; E-mail- [email protected] The cerebral cortex—the thin outer covering of the brain-is the part of the brain responsible for our ability to reason, plan, remember, and imagine. Cerebral Cortex accounts for our impressive capacity to process and transform information. The cerebral cortex is only about one-eighth of an inch thick, but it contains billions of neurons, each connected to thousands of others. The predominance of cell bodies gives the cortex a brownish gray colour. Because of its appearance, the cortex is often referred to as gray matter. Beneath the cortex are myelin-sheathed axons connecting the neurons of the cortex with those of other parts of the brain. The large concentrations of myelin make this tissue look whitish and opaque, and hence it is often referred to as white matter. The cortex is divided into two nearly symmetrical halves, the cerebral hemispheres . Thus, many of the structures of the cerebral cortex appear in both the left and right cerebral hemispheres. The two hemispheres appear to be somewhat specialized in the functions they perform. The cerebral hemispheres are folded into many ridges and grooves, which greatly increase their surface area. Each hemisphere is usually described, on the basis of the largest of these grooves or fissures, as being divided into four distinct regions or lobes. The four lobes are: • Frontal, • Parietal, • Occipital, and • Temporal. -

ARTICLES Evaluation of Spatial Working Memory Function
0031-3998/05/5806-1150 PEDIATRIC RESEARCH Vol. 58, No. 6, 2005 Copyright © 2005 International Pediatric Research Foundation, Inc. Printed in U.S.A. ARTICLES Evaluation of Spatial Working Memory Function in Children and Adults with Fetal Alcohol Spectrum Disorders: A Functional Magnetic Resonance Imaging Study KRISZTINA L. MALISZA, AVA-ANN ALLMAN, DEBORAH SHILOFF, LORNA JAKOBSON, SALLY LONGSTAFFE, AND ALBERT E. CHUDLEY National Research Council, Institute for Biodiagnostics [K.L.M., A.A.A., D.S.], Winnipeg, Manitoba, Canada, R3B 1Y6; Department of Physiology [K.L.M.], University of Manitoba, Bannatyne Campus, Winnipeg, Manitoba, Canada, R3E 3J7; Department of Psychology [L.J.], University of Manitoba, Winnipeg, Manitoba, Canada, R3T 2N2; Department of Pediatrics and Child Health [A.E.C.] and Child Development Clinic [S.L.], Children’s Hospital, Health Sciences Centre, Winnipeg, University of Manitoba, Manitoba, Canada, R3E 3P5 ABSTRACT Magnetic resonance imaging (MRI) and functional MRI stud- suggest impairment in spatial working memory in those with ies involving n-back spatial working memory (WM) tasks were FASD that does not improve with age, and that fMRI may be conducted in adults and children with Fetal Alcohol Spectrum useful in evaluation of brain function in these individuals. Disorders (FASD), and in age- and sex-matched controls. FMRI (Pediatr Res 58: 1150–1157, 2005) experiments demonstrated consistent activations in regions of the brain associated with working memory. Children with FASD displayed greater inferior-middle frontal lobe activity, while Abbreviations greater superior frontal and parietal lobe activity was observed in ARND, alcohol related neurodevelopmental disorder controls. Control children also showed an overall increase in CPT, Continuous Performance Task frontal lobe activity with increasing task difficulty, while children DLPFC, dorsolateral prefrontal cortex with FASD showed decreased activity. -

Barbey Cortex Dlpfc Working Memory.Pdf
cortex 49 (2013) 1195e1205 Available online at www.sciencedirect.com Journal homepage: www.elsevier.com/locate/cortex Research report Dorsolateral prefrontal contributions to human working memory Aron K. Barbey a,b,c,d,e,f,*,1, Michael Koenigs g and Jordan Grafman h a Decision Neuroscience Laboratory, University of Illinois at Urbana-Champaign, Champaign, IL, USA b Beckman Institute for Advanced Science and Technology, University of Illinois at Urbana-Champaign, Urbana, IL, USA c Department of Internal Medicine, University of Illinois at Urbana-Champaign, Champaign, IL, USA d Department of Psychology, University of Illinois at Urbana-Champaign, Champaign, IL, USA e Department of Speech and Hearing Science, University of Illinois at Urbana-Champaign, Champaign, IL, USA f Neuroscience Program, University of Illinois at Urbana-Champaign, Champaign, IL, USA g Department of Psychiatry, University of Wisconsin at Madison, Wisconsin’s Psychiatric Institute and Clinics, Madison, WI, USA h Traumatic Brain Injury Research Laboratory, Kessler Foundation Research Center, West Orange, NJ, USA article info abstract Article history: Although neuroscience has made remarkable progress in understanding the involvement Received 2 August 2011 of prefrontal cortex (PFC) in human memory, the necessity of dorsolateral PFC (dlPFC) for Reviewed 05 October 2011 key competencies of working memory remains largely unexplored. We therefore studied Revised 25 January 2012 human brain lesion patients to determine whether dlPFC is necessary for working memory Accepted 16 May 2012 function, administering subtests of the Wechsler Memory Scale, the Wechsler Adult Action editor Mike Kopelman Intelligence Scale, and the N-Back Task to three participant groups: dlPFC lesions (n ¼ 19), Published online 16 June 2012 non-dlPFC lesions (n ¼ 152), and no brain lesions (n ¼ 54). -

A Practical Review of Functional MRI Anatomy of the Language and Motor Systems
REVIEW ARTICLE FUNCTIONAL A Practical Review of Functional MRI Anatomy of the Language and Motor Systems X V.B. Hill, X C.Z. Cankurtaran, X B.P. Liu, X T.A. Hijaz, X M. Naidich, X A.J. Nemeth, X J. Gastala, X C. Krumpelman, X E.N. McComb, and X A.W. Korutz ABSTRACT SUMMARY: Functional MR imaging is being performed with increasing frequency in the typical neuroradiology practice; however, many readers of these studies have only a limited knowledge of the functional anatomy of the brain. This text will delineate the locations, anatomic boundaries, and functions of the cortical regions of the brain most commonly encountered in clinical practice—specifically, the regions involved in movement and language. ABBREVIATIONS: FFA ϭ fusiform face area; IPL ϭ inferior parietal lobule; PPC ϭ posterior parietal cortex; SMA ϭ supplementary motor area; VOTC ϭ ventral occipitotemporal cortex his article serves as a review of the functional areas of the brain serving to analyze spatial position and the ventral stream working Tmost commonly mapped during presurgical fMRI studies, to identify what an object is. Influenced by the dorsal and ventral specifically targeting movement and language. We have compiled stream model of vision, Hickok and Poeppel2 hypothesized a sim- what we hope is a useful, easily portable, and concise resource that ilar framework for language. In this model, the ventral stream, or can be accessible to radiologists everywhere. We begin with a re- lexical-semantic system, is involved in sound-to-meaning map- view of the language-processing system. Then we describe the pings associated with language comprehension and semantic ac- gross anatomic boundaries, organization, and function of each cess. -
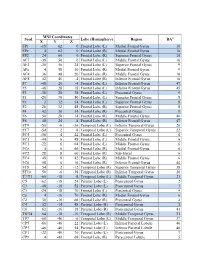
Seed MNI Coordinates Lobe
MNI Coordinates Seed Lobe (Hemisphere) Region BAa X Y Z FP1 -18 62 0 Frontal Lobe (L) Medial Frontal Gyrus 10 FPz 4 62 0 Frontal Lobe (R) Medial Frontal Gyrus 10 FP2 24 60 0 Frontal Lobe (R) Superior Frontal Gyrus 10 AF7 -38 50 0 Frontal Lobe (L) Middle Frontal Gyrus 10 AF3 -30 50 24 Frontal Lobe (L) Superior Frontal Gyrus 9 AFz 4 58 30 Frontal Lobe (R) Medial Frontal Gyrus 9 AF4 36 48 20 Frontal Lobe (R) Middle Frontal Gyrus 10 AF8 42 46 -4 Frontal Lobe (R) Inferior Frontal Gyrus 10 F7 -48 26 -4 Frontal Lobe (L) Inferior Frontal Gyrus 47 F5 -48 28 18 Frontal Lobe (L) Inferior Frontal Gyrus 45 F3 -38 28 38 Frontal Lobe (L) Precentral Gyrus 9 F1 -20 30 50 Frontal Lobe (L) Superior Frontal Gyrus 8 Fz 2 32 54 Frontal Lobe (L) Superior Frontal Gyrus 8 F2 26 32 48 Frontal Lobe (R) Superior Frontal Gyrus 8 F4 42 30 34 Frontal Lobe (R) Precentral Gyrus 9 F6 50 28 14 Frontal Lobe (R) Middle Frontal Gyrus 46 F8 48 24 -8 Frontal Lobe (R) Inferior Frontal Gyrus 47 FT9 -50 -6 -36 Temporal Lobe (L) Inferior Temporal Gyrus 20 FT7 -54 2 -8 Temporal Lobe (L) Superior Temporal Gyrus 22 FC5 -56 4 22 Frontal Lobe (L) Precentral Gyrus 6 FC3 -44 6 48 Frontal Lobe (L) Middle Frontal Gyrus 6 FC1 -22 6 64 Frontal Lobe (L) Middle Frontal Gyrus 6 FCz 4 6 66 Frontal Lobe (R) Medial Frontal Gyrus 6 FC2 28 8 60 Frontal Lobe (R) Sub-Gyral 6 FC4 48 8 42 Frontal Lobe (R) Middle Frontal Gyrus 6 FC6 58 6 16 Frontal Lobe (R) Inferior Frontal Gyrus 44 FT8 54 2 -12 Temporal Lobe (R) Superior Temporal Gyrus 38 FT10 50 -6 -38 Temporal Lobe (R) Inferior Temporal Gyrus 20 T7/T3 -

The Neuromodulatory Basis of Emotion
1 The Neuromodulatory Basis of Emotion Jean-Marc Fellous Computational Neurobiology Laboratory, The Salk Institute for Biological Studies, La Jolla, California The Neuroscientist 5(5):283-294,1999. The neural basis of emotion can be found in both the neural computation and the neuromodulation of the neural substrate mediating behavior. I review the experimental evidence showing the involvement of the hypothalamus, the amygdala and the prefrontal cortex in emotion. For each of these structures, I show the important role of various neuromodulatory systems in mediating emotional behavior. Generalizing, I suggest that behavioral complexity is partly due to the diversity and intensity of neuromodulation and hence depends on emotional contexts. Rooting the emotional state in neuromodulatory phenomena allows for its quantitative and scientific study and possibly its characterization. Key Words: Neuromodulation, Emotion, Affect, Hypothalamus, Amygdala, Prefrontal the behavior1 that this substrate mediates. The Introduction neuromodulation of 'cognitive centers' results in phenomena pertaining to emotional influences of The scientific study of the neural basis of cognitive processing. Neuromodulations of memory emotion is an active field of experimental and structures explain the influence of emotion on theoretical research (See (1,2) for reviews). Partly learning and recall; the neuromodulation of specific because of a lack of a clear definition (should it reflex pathways explains the influence of the exists) of what emotion is, and probably because of emotional state on elementary motor behaviors, and its complexity, it has been difficult to offer a so forth... neuroscience framework in which the influence of The instantaneous pattern of such modulations emotion on behavior can be studied in a (i.e. -

Frontal Lobe and Cognitive Development
Journal of Neurocytology 31, 373–385 (2002) Frontal lobe and cognitive development JOAQUIN´ M. FUSTER Neuropsychiatric Institute and Brain Research Institute, UCLA School of Medicine Los Angeles, California [email protected] Received December 1, 2002; accepted December 12, 2002 Abstract In phylogeny as in ontogeny, the association cortex of the frontal lobe, also known as the prefrontal cortex, is a late-developing region of the neocortex. It is also one of the cortical regions to undergo the greatest expansion in the course of both evolution and individual maturation. In the human adult, the prefrontal cortex constitutes as much as nearly one-third of the totality of the neocortex. The protracted, relatively large, development of the prefrontal cortex is manifest in gross morphology as well as fine structure. In the developing individual, its late maturation is made most apparent by the late myelination of its axonal connections. This and other indices of morphological development of the prefrontal cortex correlate with the development of cognitive functions that neuropsychological studies in animals and humans have ascribed to this cortex. In broad outline, the ventromedial areas of the prefrontal cortex, which with respect to other prefrontal areas develop relatively early, are involved in the expression and control of emotional and instinctual behaviors. On the other hand, the late maturing areas of the lateral prefrontal convexity are principally involved in higher executive functions. The most general executive function of the lateral prefrontal cortex is the temporal organization of goal-directed actions in the domains of behavior, cognition, and language. In all three domains, that global function is supported by a fundamental role of the lateral prefrontal cortex in temporal integration, that is, the integration of temporally discontinuous percepts and neural inputs into coherent structures of action. -

Brain Anatomy
BRAIN ANATOMY Adapted from Human Anatomy & Physiology by Marieb and Hoehn (9th ed.) The anatomy of the brain is often discussed in terms of either the embryonic scheme or the medical scheme. The embryonic scheme focuses on developmental pathways and names regions based on embryonic origins. The medical scheme focuses on the layout of the adult brain and names regions based on location and functionality. For this laboratory, we will consider the brain in terms of the medical scheme (Figure 1): Figure 1: General anatomy of the human brain Marieb & Hoehn (Human Anatomy and Physiology, 9th ed.) – Figure 12.2 CEREBRUM: Divided into two hemispheres, the cerebrum is the largest region of the human brain – the two hemispheres together account for ~ 85% of total brain mass. The cerebrum forms the superior part of the brain, covering and obscuring the diencephalon and brain stem similar to the way a mushroom cap covers the top of its stalk. Elevated ridges of tissue, called gyri (singular: gyrus), separated by shallow groves called sulci (singular: sulcus) mark nearly the entire surface of the cerebral hemispheres. Deeper groves, called fissures, separate large regions of the brain. Much of the cerebrum is involved in the processing of somatic sensory and motor information as well as all conscious thoughts and intellectual functions. The outer cortex of the cerebrum is composed of gray matter – billions of neuron cell bodies and unmyelinated axons arranged in six discrete layers. Although only 2 – 4 mm thick, this region accounts for ~ 40% of total brain mass. The inner region is composed of white matter – tracts of myelinated axons. -
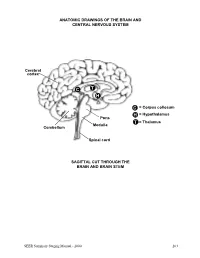
Brain and Central Nervous System
ANATOMIC DRAWINGS OF THE BRAIN AND CENTRAL NERVOUS SYSTEM Cerebral cortex C T H C = Corpus collosum H = Hypothalamus Pons T = Thalamus Medulla Cerebellum Spinal cord SAGITTAL CUT THROUGH THE BRAIN AND BRAIN STEM SEER Summary Staging Manual - 2000 263 ANATOMIC DRAWINGS OF THE BRAIN AND CENTRAL NERVOUS SYSTEM 2 1 3 4 7 5 8 6 SAGITTAL CUT THROUGH THE HUMAN HEAD WITH CEREBRUM IN PLACE The cerebrum is comprised of the: 1 Frontal lobe 2 Parietal lobe 3 Temporal lobe 4 Occipital lobe Other parts of the brain include: 5 Pons 6 Medulla (oblongata) 7 Cerebellum 8 Tentorium (cerebelli) 264 SEER Summary Staging Manual - 2000 ANATOMIC DRAWINGS OF THE BRAIN AND CENTRAL NERVOUS SYSTEM A B C D E 7 5 6 8 F SAGITTAL CUT THROUGH THE HUMAN HEAD Internal anatomy of the brain: A Inner surface of right hemisphere of cerebrum B Corpus callosum C Velum interpositum D Middle commissure E Third ventricle F Fourth ventricle Other parts of the brain (as on previous drawing): 5 Pons 6 Medulla (oblongata) 7 Cerebellum 8 Tentorium (cerebelli) SEER Summary Staging Manual - 2000 265 BRAIN AND CEREBRAL MENINGES C70.0, C71.0-C71.9 Supratentorial (S) or Infratentorial (I) C70.0 Cerebral meninges C71.0 Cerebrum ? (S) C71.1 Frontal lobe (S) C71.2 Temporal lobe (S) C71.3 Parietal lobe (S) C71.4 Occipital lobe (S) C71.5 Ventricle, NOS (S) C71.6 Cerebellum, NOS (I) C71.7 Brain stem (I) C71.8 Overlapping lesion of brain ? C71.9 Brain, NOS ? ?See Note 1. SUMMARY STAGE 1 Localized only Supratentorial tumor confined to: Cerebral hemisphere (cerebrum) or meninges of cerebral hemisphere -

Essential Neuromodulation This Page Intentionally Left Blank Essential Neuromodulation
Essential Neuromodulation This page intentionally left blank Essential Neuromodulation Jeffrey E. Arle Director, Functional Neurosurgery and Research, Department of Neurosurgery Lahey Clinic Burlington, MA Associate Professor of Neurosurgery Tufts University School of Medicine, Boston, MA Jay L. Shils Director of Intraoperative Monitoring, Dept of Neurosurgery Lahey Clinic Burlington, MA AMSTERDAM • BOSTON • HEIDELBERG • LONDON NEW YORK • OXFORD • PARIS • SAN DIEGO SAN FRANCISCO • SINGAPORE • SYDNEY • TOKYO Academic Press is an Imprint of Elsevier Academic Press is an imprint of Elsevier 32 Jamestown Road, London NW1 7BY, UK 30 Corporate Drive, Suite 400, Burlington, MA 01803, USA 525 B Street, Suite 1800, San Diego, CA 92101-4495, USA First edition 2011 Copyright © 2011 Elsevier Inc. All rights reserved No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means electronic, mechanical, photocopying, recording or otherwise without the prior written permission of the publisher Permissions may be sought directly from Elsevier's Science & Technology Rights Department in Oxford, UK: phone ( + 44) (0) 1865 843830; fax ( +44) (0) 1865 853333; email: [email protected]. Alternatively, visit the Science and Technology Books website at www.elsevierdirect.com/rights for further information Notice No responsibility is assumed by the publisher for any injury and/or damage to persons or property as a matter of products liability, negligence or otherwise, or from any use or operation of -

Frontal Lobe/Executive Functioning
Chapter 10 Frontal Lobe/Executive Functioning James G. Scott and Mike R. Schoenberg Abstract The frontal lobes represent a large area, consuming approximately one-third of the cortical surface of the brain. This area is involved directly and indirectly across a wide spectrum of human thought, behavior and emotions. The irony of the frontal lobes may best be described as the area of the brain we know the most about but understand the least. For example, frontal lobe functioning involves simple motor skills (both gross and fine), complex motor skills, sequenced motor skills, inhibition of motor skills and automatic motor skills, and these may be the simplest of the functions of the frontal lobes. The frontal lobes also subsume what is collectively referred to as executive skills. These functions include attention, rea- soning, judgment, problem solving, creativity, emotional regulation, impulse con- trol and awareness of aspects of one’s and others’ functioning. In this chapter, we will briefly discuss the anatomy of the frontal lobes, the basic and complex func- tions of the frontal lobes, and the informal assessment of frontal lobe functions. Key Points and Chapter Summary • Frontal lobes include a large area of the cortex and are involved directly or indirectly in most brain functions involving cognition, behavioral, and motor skills • Frontal lobe damage can have profound effects on attention, memory, language, problem solving/reasoning, and general comportment (person- ality/social behaviors, etc.) • Frontal lobe damage can be grouped into three syndromes determined by anatomical regions involved and associated with characteristic cognitive, (continued) J.G. Scott (*) Department of Psychiatry and Behavioral Sciences, University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA e-mail: [email protected] M.R. -

Current Understanding of the Involvement of the Insular Cortex in Neuropathic Pain: a Narrative Review
International Journal of Molecular Sciences Review Current Understanding of the Involvement of the Insular Cortex in Neuropathic Pain: A Narrative Review Ning Wang 1,2 , Yu-Han Zhang 1,2, Jin-Yan Wang 1,2,* and Fei Luo 1,2 1 CAS Key Laboratory of Mental Health, Institute of Psychology, Chinese Academy of Sciences, Beijing 100101, China; [email protected] (N.W.); [email protected] (Y.-H.Z.); [email protected] (F.L.) 2 Department of Psychology, University of Chinese Academy of Sciences, Beijing 100049, China * Correspondence: [email protected] Abstract: Neuropathic pain is difficult to cure and is often accompanied by emotional and psycho- logical changes. Exploring the mechanisms underlying neuropathic pain will help to identify a better treatment for this condition. The insular cortex is an important information integration center. Numerous imaging studies have documented increased activity of the insular cortex in the presence of neuropathic pain; however, the specific role of this region remains controversial. Early studies suggested that the insular lobe is mainly involved in the processing of the emotional motivation dimension of pain. However, increasing evidence suggests that the role of the insular cortex is more complex and may even be related to the neural plasticity, cognitive evaluation, and psychosocial aspects of neuropathic pain. These effects contribute not only to the development of neuropathic pain, but also to its comorbidity with neuropsychiatric diseases. In this review, we summarize the changes that occur in the insular cortex in the presence of neuropathic pain and analgesia, as well as the molecular mechanisms that may underlie these conditions.