Pleiotropy and the Evolution of Floral Integration
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lineage-Specific Mapping of Quantitative Trait Loci
Heredity (2013) 111, 106–113 & 2013 Macmillan Publishers Limited All rights reserved 0018-067X/13 www.nature.com/hdy ORIGINAL ARTICLE Lineage-specific mapping of quantitative trait loci C Chen1 and K Ritland We present an approach for quantitative trait locus (QTL) mapping, termed as ‘lineage-specific QTL mapping’, for inferring allelic changes of QTL evolution along with branches in a phylogeny. We describe and analyze the simplest case: by adding a third taxon into the normal procedure of QTL mapping between pairs of taxa, such inferences can be made along lineages to a presumed common ancestor. Although comparisons of QTL maps among species can identify homology of QTLs by apparent co- location, lineage-specific mapping of QTL can classify homology into (1) orthology (shared origin of QTL) versus (2) paralogy (independent origin of QTL within resolution of map distance). In this light, we present a graphical method that identifies six modes of QTL evolution in a three taxon comparison. We then apply our model to map lineage-specific QTLs for inbreeding among three taxa of yellow monkey-flower: Mimulus guttatus and two inbreeders M. platycalyx and M. micranthus, but critically assuming outcrossing was the ancestral state. The two most common modes of homology across traits were orthologous (shared ancestry of mutation for QTL alleles). The outbreeder M. guttatus had the fewest lineage-specific QTL, in accordance with the presumed ancestry of outbreeding. Extensions of lineage-specific QTL mapping to other types of data and crosses, and to inference of ancestral QTL state, are discussed. Heredity (2013) 111, 106–113; doi:10.1038/hdy.2013.24; published online 24 April 2013 Keywords: Quantitative trait locus mapping; evolution of mating systems; Mimulus INTRODUCTION common ancestor of these two taxa, back to the common ancestor of Knowledge of the genetic architecture of complex traits can offer all three taxa and forwards again to the third taxon. -

A Comprehensive Study of Metabolite Genetics Reveals Strong Pleiotropy and Heterogeneity Across Time and Context
ARTICLE https://doi.org/10.1038/s41467-019-12703-7 OPEN A comprehensive study of metabolite genetics reveals strong pleiotropy and heterogeneity across time and context Apolline Gallois1,12, Joel Mefford2,12, Arthur Ko 3, Amaury Vaysse 1, Hanna Julienne1, Mika Ala-Korpela4,5,6,7,8,9, Markku Laakso 10, Noah Zaitlen2,12*, Päivi Pajukanta 3,12*& Hugues Aschard 1,11,12* 1234567890():,; Genetic studies of metabolites have identified thousands of variants, many of which are associated with downstream metabolic and obesogenic disorders. However, these studies have relied on univariate analyses, reducing power and limiting context-specific under- standing. Here we aim to provide an integrated perspective of the genetic basis of meta- bolites by leveraging the Finnish Metabolic Syndrome In Men (METSIM) cohort, a unique genetic resource which contains metabolic measurements, mostly lipids, across distinct time points as well as information on statin usage. We increase effective sample size by an average of two-fold by applying the Covariates for Multi-phenotype Studies (CMS) approach, identifying 588 significant SNP-metabolite associations, including 228 new associations. Our analysis pinpoints a small number of master metabolic regulator genes, balancing the relative proportion of dozens of metabolite levels. We further identify associations to changes in metabolic levels across time as well as genetic interactions with statin at both the master metabolic regulator and genome-wide level. 1 Department of Computational Biology - USR 3756 CNRS, Institut Pasteur, Paris, France. 2 Department of Medicine, University of California, San Francisco, CA, USA. 3 Department of Human Genetics, University of California, Los Angeles, CA, USA. -
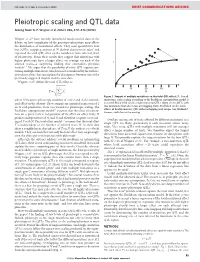
Pleiotropic Scaling and QTL Data Arising From: G
NATURE | Vol 456 | 4 December 2008 BRIEF COMMUNICATIONS ARISING Pleiotropic scaling and QTL data Arising from: G. P. Wagner et al. Nature 452, 470–473 (2008) Wagner et al.1 have recently introduced much-needed data to the debate on how complexity of the genotype–phenotype map affects the distribution of mutational effects. They used quantitative trait loci (QTLs) mapping analysis of 70 skeletal characters in mice2 and regressed the total QTL effect on the number of traits affected (level of pleiotropy). From their results they suggest that mutations with higher pleiotropy have a larger effect, on average, on each of the T affected traits—a surprising finding that contradicts previous models3–7. We argue that the possibility of some QTL regions con- taining multiple mutations, which was not considered by the authors, introduces a bias that can explain the discrepancy between one of the previously suggested models and the new data. Wagner et al.1 define the total QTL effect as vffiffiffiffiffiffiffiffiffiffiffiffiffi u uXN 10 20 30 ~t 2 T Ai N i~1 Figure 1 | Impact of multiple mutations on the total QTL effect, T. For all where N measures pleiotropy (number of traits) and Ai the normal- mutations, strict scaling according to the Euclidean superposition model is ized effect on the ith trait. They compare an empirical regression of T assumed. Black filled circles, single-mutation QTLs. Open circles, QTLs with on N with predictions from two models for pleiotropic scaling. The two mutations that affect non-overlapping traits. Red filled circles, total Euclidean superposition model3,4 assumes that the effect of a muta- effects of double-mutant QTL with overlapping trait ranges (see Methods). -

Evolution by Natural Selection, Formulated Independently by Charles Darwin and Alfred Russel Wallace
UNIT 4 EVOLUTIONARY PATT EVOLUTIONARY E RNS AND PROC E SS E Evolution by Natural S 22 Selection Natural selection In this chapter you will learn that explains how Evolution is one of the most populations become important ideas in modern biology well suited to their environments over time. The shape and by reviewing by asking by applying coloration of leafy sea The rise of What is the evidence for evolution? Evolution in action: dragons (a fish closely evolutionary thought two case studies related to seahorses) 22.1 22.4 are heritable traits that with regard to help them to hide from predators. The pattern of evolution: The process of species have changed evolution by natural and are related 22.2 selection 22.3 keeping in mind Common myths about natural selection and adaptation 22.5 his chapter is about one of the great ideas in science: the theory of evolution by natural selection, formulated independently by Charles Darwin and Alfred Russel Wallace. The theory explains how T populations—individuals of the same species that live in the same area at the same time—have come to be adapted to environments ranging from arctic tundra to tropical wet forest. It revealed one of the five key attributes of life: Populations of organisms evolve. In other words, the heritable characteris- This chapter is part of the tics of populations change over time (Chapter 1). Big Picture. See how on Evolution by natural selection is one of the best supported and most important theories in the history pages 516–517. of scientific research. -

1 ROBERT PLOMIN on Behavioral Genetics Genetic Links Common
Genetic links • Background – Why has educational psychology ben so slow ROBERT PLOMIN to accept genetics? on Behavioral Genetics • Beyond nature vs. nurture – 3 findings from genetic research with far- reaching implications for education: October, 2004 The nature of nurture The abnormal is normal Generalist genes • Molecular Genetics Quantitative genetics: twin and adoption studies Common medical disorders Identical twins Fraternal twins (monozygotic, MZ) (dizygotic, DZ) 1 Twin concordances in TEDS at 7 years Twins Early Development Study (TEDS) for low reading and maths Perceptions of nature/nurture: % parents (N=1340) and teachers Beyond nature vs. nurture: (N=667) indicating that genes are at least as important as environment (Walker & Plomin, Ed. Psych., in press) The nature of nurture • Genetic influence on experience • Genotype-environment correlation – Children’s genetic propensities are correlated with their experiences passive evocative active 2 Beyond nature vs. nurture: Genotype-environment correlation The nature of nurture • Passive • Genotype-environment correlation – e.g., family background (SES) and school – Children’s genetic propensities are correlated with achievement their experiences passive • Evocative evocative – Children evoke experiences for genetic reasons Active • Active • Molecular genetics – Children actively create environments that foster – Find genes associated with learning experiences their genetic propensities Beyond nature vs. nurture: The one-gene one-disability (OGOD) model The abnormal is normal • Common learning disabilities are the quantitative extreme of the same genetic and environmental factors responsible for normal variation in learning abilities. • There are no common learning disabilities, just the extremes of normal variation. 3 OGOD (rare) and QTL (common) disorders The quantitative trait locus (QTL) model Beyond nature vs. -

Investigations Into Roles for Endocytosis in LIN-12/Notch
Investigations into roles for endocytosis in LIN-12/Notch signaling and its regulation Jessica Chan Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy under the Executive Committee of the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2020 © 2020 Jessica Chan All Rights Reserved Abstract The LIN-12/Notch signaling pathway is highly conserved in all animals, and is crucial for proper development. It is a key pathway in specifying cell fate in many cellular contexts, and dysregulation of the pathway can have deleterious consequences. Therefore, understanding how LIN-12/Notch signaling is regulated in different contexts has been a main area of interest in the field. Previous studies in different model organisms have identified many modes of regulation of the signaling pathway, one of which is endocytosis of the ligand and receptor. Here, I further investigated the role of endocytosis in LIN-12/Notch signaling in multiple developmental contexts in Caenorhabditis elegans. Work in Drosophila and vertebrates had previously established that ligand-mediated activation of Notch requires ubiquitination of the intracellular domain of the transmembrane ligand and the activity of the endocytic adaptor Epsin in the signaling cell. The consensus in the field is that Epsin-mediated endocytosis of mono-ubiquitinated ligand generates a pulling force that exposes a cleavage site in Notch for an ADAM protease, a critical step in signal transduction. In contrast, in this thesis, I examined two different transmembrane ligands in several different cell contexts and found that activation of LIN-12/Notch and the paralogous GLP-1/Notch in C. -

Nicotinic Receptor Alpha7 Expression During Tooth Morphogenesis Reveals Functional Pleiotropy
Nicotinic Receptor Alpha7 Expression during Tooth Morphogenesis Reveals Functional Pleiotropy Scott W. Rogers1,2*, Lorise C. Gahring1,3 1 Geriatric Research, Education and Clinical Center, Veteran’s Administration, Salt Lake City, Utah, United States of America, 2 Department of Neurobiology and Anatomy, University of Utah School of Medicine, Salt Lake City, Utah, United States of America, 3 Division of Geriatrics, Department of Internal Medicine, University of Utah School of Medicine, Salt Lake City, Utah, United States of America Abstract The expression of nicotinic acetylcholine receptor (nAChR) subtype, alpha7, was investigated in the developing teeth of mice that were modified through homologous recombination to express a bi-cistronic IRES-driven tau-enhanced green fluorescent protein (GFP); alpha7GFP) or IRES-Cre (alpha7Cre). The expression of alpha7GFP was detected first in cells of the condensing mesenchyme at embryonic (E) day E13.5 where it intensifies through E14.5. This expression ends abruptly at E15.5, but was again observed in ameloblasts of incisors at E16.5 or molar ameloblasts by E17.5–E18.5. This expression remains detectable until molar enamel deposition is completed or throughout life as in the constantly erupting mouse incisors. The expression of alpha7GFP also identifies all stages of innervation of the tooth organ. Ablation of the alpha7-cell lineage using a conditional alpha7Cre6ROSA26-LoxP(diphtheria toxin A) strategy substantially reduced the mesenchyme and this corresponded with excessive epithelium overgrowth consistent with an instructive role by these cells during ectoderm patterning. However, alpha7knock-out (KO) mice exhibited normal tooth size and shape indicating that under normal conditions alpha7 expression is dispensable to this process. -

Epigenome-Wide Study Identified Methylation Sites Associated With
nutrients Article Epigenome-Wide Study Identified Methylation Sites Associated with the Risk of Obesity Majid Nikpay 1,*, Sepehr Ravati 2, Robert Dent 3 and Ruth McPherson 1,4,* 1 Ruddy Canadian Cardiovascular Genetics Centre, University of Ottawa Heart Institute, 40 Ruskin St–H4208, Ottawa, ON K1Y 4W7, Canada 2 Plastenor Technologies Company, Montreal, QC H2P 2G4, Canada; [email protected] 3 Department of Medicine, Division of Endocrinology, University of Ottawa, the Ottawa Hospital, Ottawa, ON K1Y 4E9, Canada; [email protected] 4 Atherogenomics Laboratory, University of Ottawa Heart Institute, Ottawa, ON K1Y 4W7, Canada * Correspondence: [email protected] (M.N.); [email protected] (R.M.) Abstract: Here, we performed a genome-wide search for methylation sites that contribute to the risk of obesity. We integrated methylation quantitative trait locus (mQTL) data with BMI GWAS information through a SNP-based multiomics approach to identify genomic regions where mQTLs for a methylation site co-localize with obesity risk SNPs. We then tested whether the identified site contributed to BMI through Mendelian randomization. We identified multiple methylation sites causally contributing to the risk of obesity. We validated these findings through a replication stage. By integrating expression quantitative trait locus (eQTL) data, we noted that lower methylation at cg21178254 site upstream of CCNL1 contributes to obesity by increasing the expression of this gene. Higher methylation at cg02814054 increases the risk of obesity by lowering the expression of MAST3, whereas lower methylation at cg06028605 contributes to obesity by decreasing the expression of SLC5A11. Finally, we noted that rare variants within 2p23.3 impact obesity by making the cg01884057 Citation: Nikpay, M.; Ravati, S.; Dent, R.; McPherson, R. -

QUANTITATIVE TRAIT LOCUS ANALYSIS: in Several Variants (I.E., Alleles)
TECHNOLOGIES FROM THE FIELD QUANTITATIVE TRAIT LOCUS ANALYSIS: in several variants (i.e., alleles). QTL analysis allows one MULTIPLE CROSS AND HETEROGENEOUS to map, with some precision, the genomic position of STOCK MAPPING these alleles. For many researchers in the alcohol field, the break Robert Hitzemann, Ph.D.; John K. Belknap, Ph.D.; through with respect to the genetic determination of alcohol- and Shannon K. McWeeney, Ph.D. related behaviors occurred when Plomin and colleagues (1991) made the seminal observation that a specific panel of RI mice (i.e., the BXD panel) could be used to identify KEY WORDS: Genetic theory of alcohol and other drug use; genetic the physical location of (i.e., to map) QTLs for behavioral factors; environmental factors; behavioral phenotype; behavioral phenotypes. Because the phenotypes of the different strains trait; quantitative trait gene (QTG); inbred animal strains; in this panel had been determined for many alcohol- recombinant inbred (RI) mouse strains; quantitative traits; related traits, researchers could readily apply the strategy quantitative trait locus (QTL) mapping; multiple cross mapping of RI–QTL mapping (Gora-Maslak et al. 1991). Although (MCM); heterogeneous stock (HS) mapping; microsatellite investigators recognized early on that this panel was not mapping; animal models extensive enough to answer all questions, the emerging data illustrated the rich genetic complexity of alcohol- related phenotypes (Belknap 1992; Plomin and McClearn ntil well into the 1990s, both preclinical and clinical 1993). research focused on finding “the” gene for human The next advance came with the development of diseases, including alcoholism. This focus was rein U microsatellite maps (Dietrich et al. -

An Introduction to Quantitative Genetics I Heather a Lawson Advanced Genetics Spring2018 Outline
An Introduction to Quantitative Genetics I Heather A Lawson Advanced Genetics Spring2018 Outline • What is Quantitative Genetics? • Genotypic Values and Genetic Effects • Heritability • Linkage Disequilibrium and Genome-Wide Association Quantitative Genetics • The theory of the statistical relationship between genotypic variation and phenotypic variation. 1. What is the cause of phenotypic variation in natural populations? 2. What is the genetic architecture and molecular basis of phenotypic variation in natural populations? • Genotype • The genetic constitution of an organism or cell; also refers to the specific set of alleles inherited at a locus • Phenotype • Any measureable characteristic of an individual, such as height, arm length, test score, hair color, disease status, migration of proteins or DNA in a gel, etc. Nature Versus Nurture • Is a phenotype the result of genes or the environment? • False dichotomy • If NATURE: my genes made me do it! • If NURTURE: my mother made me do it! • The features of an organisms are due to an interaction of the individual’s genotype and environment Genetic Architecture: “sum” of the genetic effects upon a phenotype, including additive,dominance and parent-of-origin effects of several genes, pleiotropy and epistasis Different genetic architectures Different effects on the phenotype Types of Traits • Monogenic traits (rare) • Discrete binary characters • Modified by genetic and environmental background • Polygenic traits (common) • Discrete (e.g. bristle number on flies) or continuous (human height) -

Synthetic Morphogenesis
Downloaded from http://cshperspectives.cshlp.org/ on September 24, 2021 - Published by Cold Spring Harbor Laboratory Press Synthetic Morphogenesis Brian P. Teague, Patrick Guye, and Ron Weiss Synthetic Biology Center, Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139 Correspondence: [email protected] Throughout biology, function is intimately linked with form. Across scales ranging from subcellular to multiorganismal, the identity and organization of a biological structure’s subunits dictate its properties. The field of molecular morphogenesis has traditionally been concerned with describing these links, decoding the molecular mechanisms that give rise to the shape and structure of cells, tissues, organs, and organisms. Recent advances in synthetic biology promise unprecedented control over these molecular mechanisms; this opens the path to not just probing morphogenesis but directing it. This review explores several frontiers in the nascent field of synthetic morphogenesis, including programmable tissues and organs, synthetic biomaterials and programmable matter, and engineering complex morphogenic systems de novo. We will discuss each frontier’s objectives, current approaches, constraints and challenges, and future potential. hat is the underlying basis of biological 2014). As the mechanistic underpinnings of Wstructure? Speculations were based on these processes are elucidated, opportunities macroscopic observation until the 17th century, arise to use this knowledge to direct mor- when Antonie van Leeuwenhoek and Robert phogenesis toward novel, useful ends. We call Hooke discovered that organisms were com- this emerging field of endeavor “synthetic mor- posed of microscopic cells (Harris 1999), and phogenesis.” Inspired by and based on natural that the size and shape of the cells affected the morphogenic systems, synthetic morphogenesis properties of the structures they formed. -

Article the Power to Detect Quantitative Trait Loci Using
The Power to Detect Quantitative Trait Loci Using Resequenced, Experimentally Evolved Populations of Diploid, Sexual Organisms James G. Baldwin-Brown,*,1 Anthony D. Long,1 and Kevin R. Thornton1 1Department of Ecology and Evolutionary Biology, University of California, Irvine *Corresponding author: E-mail: [email protected]. Associate editor: John Parsch Abstract A novel approach for dissecting complex traits is to experimentally evolve laboratory populations under a controlled environment shift, resequence the resulting populations, and identify single nucleotide polymorphisms (SNPs) and/or genomic regions highly diverged in allele frequency. To better understand the power and localization ability of such an evolve and resequence (E&R) approach, we carried out forward-in-time population genetics simulations of 1 Mb genomic regions under a large combination of experimental conditions, then attempted to detect significantly diverged SNPs. Our analysis indicates that the ability to detect differentiation between populations is primarily affected by selection coef- ficient, population size, number of replicate populations, and number of founding haplotypes. We estimate that E&R studies can detect and localize causative sites with 80% success or greater when the number of founder haplotypes is over 500, experimental populations are replicated at least 25-fold, population size is at least 1,000 diploid individuals, and the selection coefficient on the locus of interest is at least 0.1. More achievable experimental designs (less replicated, fewer founder haplotypes, smaller effective population size, and smaller selection coefficients) can have power of greater than 50% to identify a handful of SNPs of which one is likely causative. Similarly, in cases where s 0.2, less demanding experimental designs can yield high power.