Combined Transcriptome and Metabolome Analyses to Understand the Dynamic Responses of Rice Plants to Attack by the Rice Stem
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

When an Enzyme Isn't Just an Enzyme Anymore Extra Botany
eXtra Botany Insight When an enzyme isn’t just an enzyme anymore Brenda S.J. Winkel Department of Biological Sciences, Virginia Tech, Blacksburg, VA 24060, USA [email protected] In this issue (pages 1425–1440) Bross et al. provide evi- There are numerous other examples of glycolytic enzymes dence of surprising alternative functions for two iso- with alternative non-catalytic functions, though none as inten- forms of the plastid enzyme arogenate dehydratase. sively studied as GAPDH (Jeffery, 2014). The functions of This study points to a previously unsuspected con- these proteins range from serving as transcriptional regulators nection between central metabolism and chloroplast that are sensitive to sucrose concentrations to binding of plas- division and a potential new mechanism for retrograde minogen during microbial pathogenesis (Rolland et al., 2006; signaling. These findings add to a growing awareness He et al., 2013; Henderson and Martin, 2013; Vega et al., of the complexities of protein function that has sub- 2016). Dual functionality is also associated with enzymes of stantial implications for both basic and applied plant many other pathways of central metabolism, such as homoci- science. trate synthase, which is essential for lysine biosynthesis in mitochondria but can also mediate DNA repair in the nucleus We are in the midst of a paradigm shift in our understanding (Torres-Machorro et al., 2015). Aconitase is another well- of cellular metabolism, as growing numbers of proteins with established example, functioning not only as a key enzyme well-established catalytic roles are being found to have addi- in the TCA cycle, but also as an iron-responsive protein that tional, alternative functions. -

A Study of Protein Dynamics and Cofactor Interactions in Photosystem I
A STUDY OF PROTEIN DYNAMICS AND COFACTOR INTERACTIONS IN PHOTOSYSTEM I A Dissertation Presented to The Academic Faculty By Shana L. Bender In Partial Fulfillment of the Requirements for the Degree Doctorate of Philosophy in the School of Chemistry and Biochemistry Georgia Institute of Technology December 2008 A STUDY OF PROTEIN DYNAMICS AND COFACTOR INTERACTIONS IN PHOTOSYSTEM I Approved by: Dr. Bridgette Barry, Advisor Dr. Ingeborg Schmidt-Krey School of Chemistry and Biochemistry School of Biology Georgia Institute of Technology Georgia Institute of Technology Dr. Donald Doyle Dr. Nael McCarty School of Chemistry and Biochemistry Department of Pediatrics Georgia Institute of Technology Emory University Dr. Wendy Kelly School of Chemistry and Biochemistry Georgia Institute of Technology Date Approved: October 15, 2008 ACKNOWLEDGEMENTS I would like to thank my parents, who have given my strength, moral support and practical advice throughout my educational endeavors. I would also like to thank my husband who has always put my goals and education above his own ambitions. I would like to acknowledge my entire family for their constant support in my education. I would like to thank my grandparents who have kept me in their prayers, and my sister who has never doubted my career goals. I would like to acknowledge my advisor, Dr. Bridgette Barry for her valuable support throughout the years. She has been a great advisor, always challenging me to think critically. She has helped me become a better scientist. I would like to thank the past members of the Barry group: Dr. Idelisa Ayala who trained me, Dr. Colette Sacksteder who taught me to be critical of everything, and Dr. -

Chandran Et Al. Supporting Info.Pdf
Supporting Information (SI) Appendix Part 1. Impact of tissue preparation, LMD, and RNA amplification on array output. p. 2 Text S1: Detailed Experimental Design and Methods Figure S1A: Correlation analysis indicates tissue preparation has minimal impact on ATH1 array output. Figure S1B: Correlation analysis indicates RNA degradation does not significantly impact array output. Figure S1C: Correlation analysis indicates two-round amplification does not significantly impact array output. Figure S1D: Independent biological replicates of LMD samples are highly correlated. Figure S1E: Validation of LMD array expression by qPCR. Table S1: Tissue preparation-associated genes Part 2. Analysis of LMD and parallel whole leaf array data. p. 13 Table S2A: Known PM-impacted genes enriched in LMD dataset Table S2B: Dataset of LMD and whole leaf genes with PM-altered expression Table S2C: LMD PM MapMan Results and Bins Table S2D: ics1 vs. WT LMD PM MapMan Results and Bins Table S2E: Infection site-specific changes for redox and calcium categories Table S2F: cis-acting regulatory element motif analysis Part 3. Process network construction p. 149 3A. Photosynthesis 3B. Cold/dehydration response Part 4. Powdery mildew infection of WT and myb3r4 mutants p. 173 Text S4: Detailed Experimental Design and Methods (supplement to manuscript) Figure S4A. Uninfected 4 week old WT and myb3r4 plants Figure S4B. myb3r4 mutants exhibit reduced visible PM growth and reproduction Figure S4C. PM-infected WT and myb3r4 mutants do not exhibit cell death Figure S4D. Endoreduplication occurs at site of PM infection not distal to infection Figure S4E. Ploidy correlates with nuclear size. Part 1. Impact of tissue preparation, LMD, and RNA amplification on array output. -

Plastid-Localized Amino Acid Biosynthetic Pathways of Plantae Are Predominantly Composed of Non-Cyanobacterial Enzymes
Plastid-localized amino acid biosynthetic pathways of Plantae are predominantly SUBJECT AREAS: MOLECULAR EVOLUTION composed of non-cyanobacterial PHYLOGENETICS PLANT EVOLUTION enzymes PHYLOGENY Adrian Reyes-Prieto1* & Ahmed Moustafa2* Received 1 26 September 2012 Canadian Institute for Advanced Research and Department of Biology, University of New Brunswick, Fredericton, Canada, 2Department of Biology and Biotechnology Graduate Program, American University in Cairo, Egypt. Accepted 27 November 2012 Studies of photosynthetic eukaryotes have revealed that the evolution of plastids from cyanobacteria Published involved the recruitment of non-cyanobacterial proteins. Our phylogenetic survey of .100 Arabidopsis 11 December 2012 nuclear-encoded plastid enzymes involved in amino acid biosynthesis identified only 21 unambiguous cyanobacterial-derived proteins. Some of the several non-cyanobacterial plastid enzymes have a shared phylogenetic origin in the three Plantae lineages. We hypothesize that during the evolution of plastids some enzymes encoded in the host nuclear genome were mistargeted into the plastid. Then, the activity of those Correspondence and foreign enzymes was sustained by both the plastid metabolites and interactions with the native requests for materials cyanobacterial enzymes. Some of the novel enzymatic activities were favored by selective compartmentation should be addressed to of additional complementary enzymes. The mosaic phylogenetic composition of the plastid amino acid A.R.-P. ([email protected]) biosynthetic pathways and the reduced number of plastid-encoded proteins of non-cyanobacterial origin suggest that enzyme recruitment underlies the recompartmentation of metabolic routes during the evolution of plastids. * Equal contribution made by these authors. rimary plastids of plants and algae are the evolutionary outcome of an endosymbiotic association between eukaryotes and cyanobacteria1. -

Characterization of Six Arabidopsis AROGENATE DEHYDRATASE Promoters
Western University Scholarship@Western Electronic Thesis and Dissertation Repository 11-3-2017 10:00 AM Characterization of six Arabidopsis AROGENATE DEHYDRATASE promoters Emily J. Cornelius The University of Western Ontario Supervisor Dr. Susanne E. Kohalmi The University of Western Ontario Graduate Program in Biology A thesis submitted in partial fulfillment of the equirr ements for the degree in Master of Science © Emily J. Cornelius 2017 Follow this and additional works at: https://ir.lib.uwo.ca/etd Part of the Biology Commons Recommended Citation Cornelius, Emily J., "Characterization of six Arabidopsis AROGENATE DEHYDRATASE promoters" (2017). Electronic Thesis and Dissertation Repository. 5081. https://ir.lib.uwo.ca/etd/5081 This Dissertation/Thesis is brought to you for free and open access by Scholarship@Western. It has been accepted for inclusion in Electronic Thesis and Dissertation Repository by an authorized administrator of Scholarship@Western. For more information, please contact [email protected]. Abstract Phenylalanine is an important aromatic amino acid synthesized by higher plants, and is a major component of numerous specialized metabolites including structural components, pigments, and defense compounds. The last step in the synthesis of phenylalanine is catalyzed by an enzyme called AROGENATE DEHYDRATASE, of which there are six different isoenzymes encoded by the Arabidopsis genome. All six have specialized roles within the plant, and are differentially expressed during development and under stressful conditions. To deduce the potential specialized role of each ADT, unique patterns of regulatory motifs were identified for all six ADT promoters, as well as corresponding transcription factors with similar expression profiles to each enzyme. Seven stable transgenic Arabidopsis lines were also generated using ADT promoter-eGFP/GUS constructs to test expression in all tissues during development, and under stressful conditions. -

When an Enzyme Isn't Just an Enzyme Anymore Extra Botany
eXtra Botany Insight When an enzyme isn’t just an enzyme anymore Brenda S.J. Winkel Downloaded from https://academic.oup.com/jxb/article-abstract/68/7/1387/3778342/ by University Libraries | Virginia Tech user on 07 October 2019 Department of Biological Sciences, Virginia Tech, Blacksburg, VA 24060, USA [email protected] In this issue (pages 1425–1440) Bross et al. provide evi- There are numerous other examples of glycolytic enzymes dence of surprising alternative functions for two iso- with alternative non-catalytic functions, though none as inten- forms of the plastid enzyme arogenate dehydratase. sively studied as GAPDH (Jeffery, 2014). The functions of This study points to a previously unsuspected con- these proteins range from serving as transcriptional regulators nection between central metabolism and chloroplast that are sensitive to sucrose concentrations to binding of plas- division and a potential new mechanism for retrograde minogen during microbial pathogenesis (Rolland et al., 2006; signaling. These findings add to a growing awareness He et al., 2013; Henderson and Martin, 2013; Vega et al., of the complexities of protein function that has sub- 2016). Dual functionality is also associated with enzymes of stantial implications for both basic and applied plant many other pathways of central metabolism, such as homoci- science. trate synthase, which is essential for lysine biosynthesis in mitochondria but can also mediate DNA repair in the nucleus We are in the midst of a paradigm shift in our understanding (Torres-Machorro et al., 2015). Aconitase is another well- of cellular metabolism, as growing numbers of proteins with established example, functioning not only as a key enzyme well-established catalytic roles are being found to have addi- in the TCA cycle, but also as an iron-responsive protein that tional, alternative functions. -

The Effect of Metal Composition and Particle Size on Nanostructure-Toxicity in Plants
BearWorks MSU Graduate Theses Summer 2019 The Effect of Metal Composition and Particle Size on Nanostructure-Toxicity in Plants Natalie Lynn Smith Missouri State University, [email protected] As with any intellectual project, the content and views expressed in this thesis may be considered objectionable by some readers. However, this student-scholar’s work has been judged to have academic value by the student’s thesis committee members trained in the discipline. The content and views expressed in this thesis are those of the student-scholar and are not endorsed by Missouri State University, its Graduate College, or its employees. Follow this and additional works at: https://bearworks.missouristate.edu/theses Part of the Biology Commons, and the Genetics and Genomics Commons Recommended Citation Smith, Natalie Lynn, "The Effect of Metal Composition and Particle Size on Nanostructure-Toxicity in Plants" (2019). MSU Graduate Theses. 3429. https://bearworks.missouristate.edu/theses/3429 This article or document was made available through BearWorks, the institutional repository of Missouri State University. The work contained in it may be protected by copyright and require permission of the copyright holder for reuse or redistribution. For more information, please contact [email protected]. THE EFFECT OF METAL COMPOSITION AND PARTICLE SIZE ON NANOSTRUCTURE-TOXICITY IN PLANTS A Master’s Thesis Presented to The Graduate College of Missouri State University TEMPLATE In Partial Fulfillment Of the Requirements for the Degree Master of Science, Biology By Natalie Lynn Smith August 2019 Copyright 2019 by Natalie Lynn Smith ii THE EFFECT OF METAL COMPOSITION AND PARTICLE SIZE ON NANOSTRUCTURE-TOXICITY IN PLANTS Biology Missouri State University, August 2019 Master of Science Natalie Lynn Smith ABSTRACT Silver nanoparticles (AgNPs) have consistently been shown to have a detrimental effect on bacteria, fungi, and plants. -

Complex Evolutionary Events at a Tandem Cluster of Arabidopsis Thaliana Genes Resulting in a Single- Locus Genetic Incompatibility
Complex Evolutionary Events at a Tandem Cluster of Arabidopsis thaliana Genes Resulting in a Single- Locus Genetic Incompatibility The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Smith, Lisa M., Kirsten Bomblies, and Detlef Weigel. 2011. Complex evolutionary events at a tandem cluster of Arabidopsis thaliana genes resulting in a single-locus genetic incompatibility. PLoS Genetics 7(7): e1002164. Published Version doi:10.1371/journal.pgen.1002164 Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:9976281 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Open Access Policy Articles, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#OAP Complex Evolutionary Events at a Tandem Cluster of Arabidopsis thaliana Genes Resulting in a Single-Locus Genetic Incompatibility Lisa M. Smith, Kirsten Bomblies¤, Detlef Weigel* Department of Molecular Biology, Max Planck Institute for Developmental Biology, Tu¨bingen, Germany Abstract Non-additive interactions between genomes have important implications, not only for practical applications such as breeding, but also for understanding evolution. In extreme cases, genes from different genomic backgrounds may be incompatible and compromise normal development or physiology. Of particular interest are non-additive interactions of alleles at the same locus. For example, overdominant behavior of alleles, with respect to plant fitness, has been proposed as an important component of hybrid vigor, while underdominance may lead to reproductive isolation. Despite their importance, only a few cases of genetic over- or underdominance affecting plant growth or fitness are understood at the level of individual genes. -

Vhe Aromatic Amino Acid Pathway Branches at L-Arogenate In,Uglena Gracilisl7j3, GRAHAM S
MOLECULAR AND CELLULAR BIOLOGY, May 1981, p. 426-438 Vol. 1, No. 5 0270-7306/81/050426-13$2.00/0 )Vhe Aromatic Amino Acid Pathway Branches at L-Arogenate in,uglena gracilisL7j3, GRAHAM S. BYNG, ROBERT J.JWHITAKER, CHARLES L.(SHAPIRO, AND ROY A.4:NSEN* Center for Soma c-Cell Genetics and Biochemistry, Department of Biological Sciences, State University of New York at Binghamton, Binghamton, New York 13901 Received 5 December 1980/Accepted 6 March 1981 The recently characterized amino acid L-arogenate (Zamir et al., J. Am. Chem. Soc. 102:4499-4504, 1980) may be a precursor of either L-phenylalanine or L- tyrosine in nature. Euglena gracilis is the first example of an organism that uses L-arogenate as the sole precursor of both L-tyrosine and L-phenylalanine, thereby creating a pathway in which L-arogenate rather than prephenate becomes the metabolic branch point. E. gracilis ATCC 12796 was cultured in the light under myxotrophic conditions and harvested in late exponential phase before extract preparation for enzymological assays. Arogenate dehydrogenase was dependent upon nicotinamide adenine dinucleotide phosphate for activity. L-Tyrosine in- hibited activity effectively with kinetics that were competitive with respect to L- arogenate and noncompetitive with respect to nicotinamide adenine dinucleotide phosphate. The possible inhibition of arogenate dehydratase by L-phenylalanine has not yet been determined. Beyond the latter uncertainty, the overall regulation of aromatic biosynthesis was studied through the characterization of 3-deoxy-D- arabino-heptulosonate 7-phosphate synthase and chorismate mutase. 3-Deoxy- D-arabino-heptulosonate 7-phosphate synthase was subject to noncompetitive inhibition by L-tyrosine with respect to either of the two substrates. -

Phenylalanine Biosynthesis and Its Relationship to Accumulation of Capsaicinoids During Capsicum Chinense Fruit Development
DOI: 10.1007/s10535-016-0608-4 BIOLOGIA PLANTARUM 60 (3): 579-584, 2016 Phenylalanine biosynthesis and its relationship to accumulation of capsaicinoids during Capsicum chinense fruit development L.A. CASTRO-CONCHA, F.M. BAAS-ESPINOLA, W.R. ANCONA-ESCALANTE, F.A. VÁZQUEZ-FLOTA, and M.L. MIRANDA-HAM* Unidad de Bioquímica y Biología Molecular de Plantas, Centro de Investigación Científica de Yucatán, Mérida, 97200, Yucatán, México Abstract Activities of phenylalanine (Phe) biosynthetic enzymes chorismate mutase (CM) and arogenate dehydratase (ADT) and of phenylalanine ammonia lyase [PAL, an enzyme that directs Phe towards capsaicinoid (CAP) synthesis] were analyzed during Capsicum chinense Jacq. (habanero pepper) fruit development. A maximum CM activity coincided with a maximum CAP accumulation. However, ADT exhibited two activity peaks, one during the early phase (10 - 17 days post-anthesis, DPA) and another during the late phase (35 - 37 DPA); only the latter coincided with CAP. Interestingly, PAL activity was inversely related to CAP accumulation; lower activities coincided with a maximum CAP content. These results suggest the operation of a control mechanism that coordinated Phe synthesis and its channeling towards CAP synthesis during the course of fruit development. Additional key words: arogenate dehydratase, chorismate mutase, fruit placenta, habanero pepper, phenylalanine ammonia lyase. Introduction The burning sensation that is typical of hot peppers is accumulation of CAP and their intermediates suggesting caused by capsaicinoids (CAP), a group of arylamides, that a higher Phe supply might be channeled to CAP containing a vanilloid moiety and an acyl side chain, that synthesis (Salgado-Garciglia and Ochoa-Alejo 1990, are derived from phenylalanine (Phe) and a branched Ochoa-Alejo and Salgado-Garciglia 1992). -
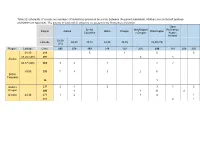
Table S1: Schematic of Crosses and Numbers of Individuals Produced by a Cross Between the Parent Individuals
Table S1: Schematic of crosses and numbers of individuals produced by a cross between the parent individuals. Mothers are on the left (yellow) and fathers on top (blue). The parents in bold and all offspring are present in the Krusenberg field trial. Open British Washington Pollination, Region Alaska Idaho Oregon Washington Columbia x Oregon Koster, Holland 58,30 Latitude 60,00 49,15 48,25 45,35 45,50 (75) (15) Region Latitude Clone 293 278 193 214 159 236 299 187 209 290 58,30 216 5 1 5 5 59,20 (500) 291 2 1 Alaska 60,37 (400) 265 3 3 1 2 2 49,00 230 7 4 3 2 6 British Columbia 96 1 Idaho x 237 2 1 3 3 1 3 Oregon 240 4 1 21 2 Oregon 44,30 271 1 2 1 4 1 301 2 1 Table S2: Monthly mean temperature and rainfall information for years 2017 and 2018. 2017 2018 Temp. (oC) Rainfall Temp. (oC) Rainfall (mm) (mm) Jan -1.6 15.8 -1.4 53.6 Feb -1.14 19.2 -4.56 19.1 Mar 2.25 28.5 -3.11 23.1 Apr 3.76 26.2 6.05 36.3 May 10.45 10.4 15.33 6.7 Jun 14.7 50.6 16.3 20.7 Jul 16.62 14.6 21.6 81.7 Aug 15.78 61.1 17.77 68.7 Sep 12.16 68.2 12.86 42.2 Oct 6.79 89.9 7.48 21.1 Nov 2.61 70.5 3.41 24.3 Dec 0.18 49.9 -0.28 31.1 Table S3: Drawing, picture and description of all 6 stages of bud burst. -

Multiscale Metabolic Modeling of C4 Plants: Connecting Nonlinear Genome-Scale Models to Leaf-Scale Metabolism in Developing Maize Leaves
MULTISCALE METABOLIC MODELING OF C4 PLANTS: CONNECTING NONLINEAR GENOME-SCALE MODELS TO LEAF-SCALE METABOLISM IN DEVELOPING MAIZE LEAVES ELI BOGART AND CHRISTOPHER R. MYERS Abstract. C4 plants, such as maize, concentrate carbon dioxide in a special- ized compartment surrounding the veins of their leaves to improve the efficiency of carbon dioxide assimilation. Nonlinear relationships between carbon diox- ide and oxygen levels and reaction rates are key to their physiology but cannot be handled with standard techniques of constraint-based metabolic modeling. We demonstrate that incorporating these relationships as constraints on re- action rates and solving the resulting nonlinear optimization problem yields realistic predictions of the response of C4 systems to environmental and bio- chemical perturbations. Using a new genome-scale reconstruction of maize metabolism, we build an 18000-reaction, nonlinearly constrained model de- scribing mesophyll and bundle sheath cells in 15 segments of the developing maize leaf, interacting via metabolite exchange, and use RNA-seq and en- zyme activity measurements to predict spatial variation in metabolic state by a novel method that optimizes correlation between fluxes and expression data. Though such correlations are known to be weak in general, here the predicted fluxes achieve high correlation with the data, successfully capture the experi- mentally observed base-to-tip transition between carbon-importing tissue and carbon-exporting tissue, and include a nonzero growth rate, in contrast to prior results