Distillation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Liquid-Liquid Extraction
OCTOBER 2020 www.processingmagazine.com A BASIC PRIMER ON LIQUID-LIQUID EXTRACTION IMPROVING RELIABILITY IN CHEMICAL PROCESSING WITH PREVENTIVE MAINTENANCE DRIVING PACKAGING SUSTAINABILITY IN THE TIME OF COVID-19 Detecting & Preventing Pressure Gauge AUTOMATIC RECIRCULATION VALVES Failures Schroedahl www.circor.com/schroedahl page 48 LOW-PROFILE, HIGH-CAPACITY SCREENER Kason Corporation www.kason.com page 16 chemical processing A basic primer on liquid-liquid extraction An introduction to LLE and agitated LLE columns | By Don Glatz and Brendan Cross, Koch Modular hemical engineers are often faced with The basics of liquid-liquid extraction the task to design challenging separation While distillation drives the separation of chemicals C processes for product recovery or puri- based upon dif erences in relative volatility, LLE is a fication. This article looks at the basics separation technology that exploits the dif erences in of one powerful and yet overlooked separation tech- the relative solubilities of compounds in two immis- nique: liquid-liquid extraction. h ere are other unit cible liquids. Typically, one liquid is aqueous, and the operations used to separate compounds, such as other liquid is an organic compound. distillation, which is taught extensively in chemical Used in multiple industries including chemical, engineering curriculums. If a separation is feasible by pharmaceutical, petrochemical, biobased chemicals distillation and is economical, there is no reason to and l avor and fragrances, this approach takes careful consider liquid-liquid extraction (LLE). However, dis- process design by experienced chemical engineers and tillation may not be a feasible solution for a number scientists. In many cases, LLE is the best choice as a of reasons, such as: separation technology and well worth searching for a • If it requires a complex process sequence (several quali ed team to assist in its development and design. -

Selection of Thermodynamic Methods
P & I Design Ltd Process Instrumentation Consultancy & Design 2 Reed Street, Gladstone Industrial Estate, Thornaby, TS17 7AF, United Kingdom. Tel. +44 (0) 1642 617444 Fax. +44 (0) 1642 616447 Web Site: www.pidesign.co.uk PROCESS MODELLING SELECTION OF THERMODYNAMIC METHODS by John E. Edwards [email protected] MNL031B 10/08 PAGE 1 OF 38 Process Modelling Selection of Thermodynamic Methods Contents 1.0 Introduction 2.0 Thermodynamic Fundamentals 2.1 Thermodynamic Energies 2.2 Gibbs Phase Rule 2.3 Enthalpy 2.4 Thermodynamics of Real Processes 3.0 System Phases 3.1 Single Phase Gas 3.2 Liquid Phase 3.3 Vapour liquid equilibrium 4.0 Chemical Reactions 4.1 Reaction Chemistry 4.2 Reaction Chemistry Applied 5.0 Summary Appendices I Enthalpy Calculations in CHEMCAD II Thermodynamic Model Synopsis – Vapor Liquid Equilibrium III Thermodynamic Model Selection – Application Tables IV K Model – Henry’s Law Review V Inert Gases and Infinitely Dilute Solutions VI Post Combustion Carbon Capture Thermodynamics VII Thermodynamic Guidance Note VIII Prediction of Physical Properties Figures 1 Ideal Solution Txy Diagram 2 Enthalpy Isobar 3 Thermodynamic Phases 4 van der Waals Equation of State 5 Relative Volatility in VLE Diagram 6 Azeotrope γ Value in VLE Diagram 7 VLE Diagram and Convergence Effects 8 CHEMCAD K and H Values Wizard 9 Thermodynamic Model Decision Tree 10 K Value and Enthalpy Models Selection Basis PAGE 2 OF 38 MNL 031B Issued November 2008, Prepared by J.E.Edwards of P & I Design Ltd, Teesside, UK www.pidesign.co.uk Process Modelling Selection of Thermodynamic Methods References 1. -

Distillation Theory
Chapter 2 Distillation Theory by Ivar J. Halvorsen and Sigurd Skogestad Norwegian University of Science and Technology Department of Chemical Engineering 7491 Trondheim, Norway This is a revised version of an article published in the Encyclopedia of Separation Science by Aca- demic Press Ltd. (2000). The article gives some of the basics of distillation theory and its purpose is to provide basic understanding and some tools for simple hand calculations of distillation col- umns. The methods presented here can be used to obtain simple estimates and to check more rigorous computations. NTNU Dr. ing. Thesis 2001:43 Ivar J. Halvorsen 28 2.1 Introduction Distillation is a very old separation technology for separating liquid mixtures that can be traced back to the chemists in Alexandria in the first century A.D. Today distillation is the most important industrial separation technology. It is particu- larly well suited for high purity separations since any degree of separation can be obtained with a fixed energy consumption by increasing the number of equilib- rium stages. To describe the degree of separation between two components in a column or in a column section, we introduce the separation factor: ()⁄ xL xH S = ------------------------T (2.1) ()x ⁄ x L H B where x denotes mole fraction of a component, subscript L denotes light compo- nent, H heavy component, T denotes the top of the section, and B the bottom. It is relatively straightforward to derive models of distillation columns based on almost any degree of detail, and also to use such models to simulate the behaviour on a computer. -

Distillation Accessscience from McgrawHill Education
6/19/2017 Distillation AccessScience from McGrawHill Education (http://www.accessscience.com/) Distillation Article by: King, C. Judson University of California, Berkeley, California. Last updated: 2014 DOI: https://doi.org/10.1036/10978542.201100 (https://doi.org/10.1036/10978542.201100) Content Hide Simple distillations Fractional distillation Vaporliquid equilibria Distillation pressure Molecular distillation Extractive and azeotropic distillation Enhancing energy efficiency Computational methods Stage efficiency Links to Primary Literature Additional Readings A method for separating homogeneous mixtures based upon equilibration of liquid and vapor phases. Substances that differ in volatility appear in different proportions in vapor and liquid phases at equilibrium with one another. Thus, vaporizing part of a volatile liquid produces vapor and liquid products that differ in composition. This outcome constitutes a separation among the components in the original liquid. Through appropriate configurations of repeated vaporliquid contactings, the degree of separation among components differing in volatility can be increased manyfold. See also: Phase equilibrium (/content/phaseequilibrium/505500) Distillation is by far the most common method of separation in the petroleum, natural gas, and petrochemical industries. Its many applications in other industries include air fractionation, solvent recovery and recycling, separation of light isotopes such as hydrogen and deuterium, and production of alcoholic beverages, flavors, fatty acids, and food oils. Simple distillations The two most elementary forms of distillation are a continuous equilibrium distillation and a simple batch distillation (Fig. 1). http://www.accessscience.com/content/distillation/201100 1/10 6/19/2017 Distillation AccessScience from McGrawHill Education Fig. 1 Simple distillations. (a) Continuous equilibrium distillation. -

Distillation Design the Mccabe-Thiele Method Distiller Diagam Introduction
Distillation Design The McCabe-Thiele Method Distiller diagam Introduction • UiUsing r igorous tray-bby-tray callilculations is time consuming, and is often unnecessary. • OikhdfiifbOne quick method of estimation for number of plates and feed stage can be obtained from the graphical McCabe-Thiele Method. • This eliminates the need for tedious calculations, and is also the first step to understanding the Fenske-Underwood- Gilliland method for multi-component distillation • Typi ica lly, the in le t flow to the dis tilla tion co lumn is known, as well as mole percentages to feed plate, because these would be specified by plant conditions. • The desired composition of the bottoms and distillate prodillbifiddhiilldducts will be specified, and the engineer will need to design a distillation column to produce these results. • With the McCabe-Thiele Method, the total number of necessary plates, as well as the feed plate location can biddbe estimated, and some ifiinformation can a lblso be determined about the enthalpic condition of the feed and reflux ratio. • This method assumes that the column is operated under constant pressure, and the constant molal overflow assumption is necessary, which states that the flow rates of liquid and vapor do not change throughout the column. • To understand this method, it is necessary to first elaborate on the subjects of the x-y diagram, and the operating lines used to create the McCabe-Thiele diagram. The x-y diagram • The x-y diagram depi icts vapor-liliqu id equilibrium data, where any point on the curve shows the variations of the amount of liquid that is in equilibrium with vapor at different temperatures. -
Minimum Reflux Ratio
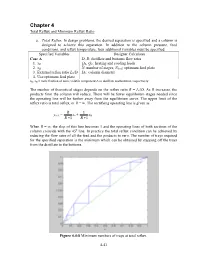
Chapter 4 Total Reflux and Minimum Reflux Ratio a. Total Reflux . In design problems, the desired separation is specified and a column is designed to achieve this separation. In addition to the column pressure, feed conditions, and reflux temperature, four additional variables must be specified. Specified Variables Designer Calculates Case A D, B: distillate and bottoms flow rates 1. xD QR, QC: heating and cooling loads 2. xB N: number of stages, Nfeed : optimum feed plate 3. External reflux ratio L0/D DC: column diameter 4. Use optimum feed plate xD, xB = mole fraction of more volatile component A in distillate and bottoms, respectively The number of theoretical stages depends on the reflux ratio R = L0/D. As R increases, the products from the column will reduce. There will be fewer equilibrium stages needed since the operating line will be further away from the equilibrium curve. The upper limit of the reflux ratio is total reflux, or R = ∞. The rectifying operating line is given as R 1 yn+1 = xn + xD R +1 R +1 When R = ∞, the slop of this line becomes 1 and the operating lines of both sections of the column coincide with the 45 o line. In practice the total reflux condition can be achieved by reducing the flow rates of all the feed and the products to zero. The number of trays required for the specified separation is the minimum which can be obtained by stepping off the trays from the distillate to the bottoms. Figure 4.4-8 Minimum numbers of trays at total reflux. -

I. an Improved Procedure for Alkene Ozonolysis. II. Exploring a New Structural Paradigm for Peroxide Antimalarials
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Student Research Projects, Dissertations, and Theses - Chemistry Department Chemistry, Department of 2011 I. An Improved Procedure for Alkene Ozonolysis. II. Exploring a New Structural Paradigm for Peroxide Antimalarials. Charles Edward Schiaffo University of Nebraska-Lincoln Follow this and additional works at: https://digitalcommons.unl.edu/chemistrydiss Part of the Organic Chemistry Commons Schiaffo, Charles Edward, "I. An Improved Procedure for Alkene Ozonolysis. II. Exploring a New Structural Paradigm for Peroxide Antimalarials." (2011). Student Research Projects, Dissertations, and Theses - Chemistry Department. 23. https://digitalcommons.unl.edu/chemistrydiss/23 This Article is brought to you for free and open access by the Chemistry, Department of at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Student Research Projects, Dissertations, and Theses - Chemistry Department by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. I. An Improved Procedure for Alkene Ozonolysis. II. Exploring a New Structural Paradigm for Peroxide Antimalarials. By Charles E. Schiaffo A DISSERTATION Presented to the Faculty of The Graduate College at the University of Nebraska In Partial Fulfillment of Requirements For the Degree of Doctor of Philosophy Major: Chemistry Under the Supervision of Professor Patrick H. Dussault Lincoln, Nebraska June, 2011 I. An Improved Procedure for Alkene Ozonolysis. II. Exploring a New Structural Paradigm for Peroxide Antimalarials. Charles E. Schiaffo, Ph.D. University of Nebraska-Lincoln, 2011 Advisor: Patrick H. Dussault The use of ozone for the transformation of alkenes to carbonyls has been well established. The reaction of ozone with alkenes in this fashion generates either a 1,2,4- trioxolane (ozonide) or a hydroperoxyacetal, either of which must undergo a separate reduction step to provide the desired carbonyl compound. -

Effect of Pressure on Distillation Separation Operation
Solutions for R&D to Design PreFEED Effect of Pressure on Distillation Separation Operation April 30, 2012 PreFEED Corporation Hiromasa Taguchi Solutions for R&D to Design 1 PreFEED Introduction Generally, it is known that, for distillation, separation tends to be enhanced by lower pressures. For two components, the ease of distillation separation can be judged from the value of the relative volatility (α). Here, we will take typical substances as examples and examine the effect of pressure changes on their relative volatilities. Solutions for R&D to Design 2 PreFEED Ideal Solution System The relative volatility (α) is a ratio of vapor-liquid equilibrium ratios and can be expressed by the following equation: K1 y1 y2 K1 , K2 K2 x1 x2 In the case of an ideal solution system , using Raoult’s law, the relative volatility can be expressed as a ratio of vapor pressures. Py x Po 1 1 1 Raoult’s law Py x Po o 2 2 2 K1 P1 y Po o K 1 1 K2 P2 1 x1 P o y2 P2 K2 x2 P Solutions for R&D to Design 3 PreFEED Methanol-Ethanol System As an example of an ideal solution system , let’s consider a methanol-ethanol binary system. α is calculated by obtaining vapor pressures from the Antoine constants in the table below. 0 B 2.8 ln(Pi [Pa]) A 2.6 C T[K] 2.4 ABC 2.2 Methanol 23.4803 3626.55 -34.29 2 Ethanol 23.8047 3803.98 -41.68 1.8 As the temperature (saturated 1.6 pressure) decreases, the value of 1.4 α increases. -

Acid Dissociation Constant - Wikipedia, the Free Encyclopedia Page 1
Acid dissociation constant - Wikipedia, the free encyclopedia Page 1 Help us provide free content to the world by donating today ! Acid dissociation constant From Wikipedia, the free encyclopedia An acid dissociation constant (aka acidity constant, acid-ionization constant) is an equilibrium constant for the dissociation of an acid. It is denoted by Ka. For an equilibrium between a generic acid, HA, and − its conjugate base, A , The weak acid acetic acid donates a proton to water in an equilibrium reaction to give the acetate ion and − + HA A + H the hydronium ion. Key: Hydrogen is white, oxygen is red, carbon is gray. Lines are chemical bonds. K is defined, subject to certain conditions, as a where [HA], [A−] and [H+] are equilibrium concentrations of the reactants. The term acid dissociation constant is also used for pKa, which is equal to −log 10 Ka. The term pKb is used in relation to bases, though pKb has faded from modern use due to the easy relationship available between the strength of an acid and the strength of its conjugate base. Though discussions of this topic typically assume water as the solvent, particularly at introductory levels, the Brønsted–Lowry acid-base theory is versatile enough that acidic behavior can now be characterized even in non-aqueous solutions. The value of pK indicates the strength of an acid: the larger the value the weaker the acid. In aqueous a solution, simple acids are partially dissociated to an appreciable extent in in the pH range pK ± 2. The a actual extent of the dissociation can be calculated if the acid concentration and pH are known. -

AP42 Chapter 9 Reference
Background Report Reference AP-42 Section Number: 9.2.2 Background Chapter: 4 Reference Number: 22 Title: "Critical Review of Henry's Law Constants fro Pesticides" in Reviews of Environmental Contamination and Toxicology L.R. Suntio 1988 AP-42 Section 9! Reference Report Sect. 3c L2% Critical Review of Henry's Law Constants Reference for Pesticides L.R. Sunti0,'W.Y. Shiu,* D. Mackay,* J.N. Seiber,** and D. Glotfelty*** Contents ......... ........... ........... ............. IV. Data Analysis ........ ........... ......... I V. Discussion .......... ........... .......... 41 ............. so .......... ............ I. Introduction Pesticides play an important role in maintaining agricultural productivity, but they may also be causes of contamination of air, water, soil, and food, with possible adverse effects on human and animal health. The proper use of pesticide chemicals must be based on an understanding of the behavior of the chemicals as they interact with air, water, soil, and biota, react or degrade, and migrate. This behavior is strongly influenced by the chemicals' physical- chemical properties of solubility in water, vapor pressure or volatility, and tendency to sorb to organic and mineral matter in the soil. Reviews of such physical-chemical properties have been compiled by Kenaga (1980), Kenaga and Goring (1980), Briggs (1981), and Bowman and Sans (1983) for aqueous solubility, octanol-water partition coefficient, bio- accumulation, and soil sorption; Spencer and Cliath (1970, 1973. 1983). and Spencer (1976) for vapor pressure and volatilization from soil. In this chapter we compile and discuss data for Henry's Law constant H (which is the ratio of solute partial pressure in the air to the equilibrium water concentration and thus has units of Pa m3/mol) or the air-water partition 'Department ofchemical Engineering and Applied Chemistry. -

Energy Efficient Trace Removal by Extractive Distillation
Energy efficient trace removal by extractive distillation Citation for published version (APA): Jongmans, M. T. G. (2012). Energy efficient trace removal by extractive distillation. Technische Universiteit Eindhoven. https://doi.org/10.6100/IR732573 DOI: 10.6100/IR732573 Document status and date: Published: 01/01/2012 Document Version: Publisher’s PDF, also known as Version of Record (includes final page, issue and volume numbers) Please check the document version of this publication: • A submitted manuscript is the version of the article upon submission and before peer-review. There can be important differences between the submitted version and the official published version of record. People interested in the research are advised to contact the author for the final version of the publication, or visit the DOI to the publisher's website. • The final author version and the galley proof are versions of the publication after peer review. • The final published version features the final layout of the paper including the volume, issue and page numbers. Link to publication General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal. -

A Study of the Reversing of Relative Volatilities by Extractive Distillation
A study of the reversing of relative volatilities by extractive distillation by An-I Yeh A thesis submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemical Engineering Montana State University © Copyright by An-I Yeh (1986) Abstract: The separation of the close-boiling mixture, m-xylene-o-xylene; three binary azeotropes: ethanol-water, methyl acetate-methanol, acetone-methanol, and four ternary azeotropes: n-propyl acetate-n-propanol-water, isopropyl acetate-isopropanol-water, n—butyl acetate-n- butanol-water, isobutyl acetate-isobutanol-water has been enhanced by extractive distillation. The azeotropes have been negated and the relative volatilities of key components have been reversed by the agents used. The plot of polar interaction versus hydrogen bonding, called polarity diagram, was used to compare the affinity of agents for key components. Thus the key component which will be the overhead product can be predicted. The three solubility parameters were used to describe the intermolecular forces occurring between agents and key components in extractive distillation. The MOSCED model was used to calculate the activity coefficients of the key components using the properties of the pure compounds. The calculated values fitted the experimental data well. The advantage of this model was to calculate the. relative volatilities of key components in the presence of the agent using the properties of pure compounds instead of using the properties of mixtures. Temperature inversion, where the overhead temperature was higher than the stillpot temperature, was observed for the acetone-methanol system when ketones were used as the agents. The data showed that the temperature inversion could be caused by the dissolving of the vapor of key components in the liquid agents.