HH-Suite User Guide
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
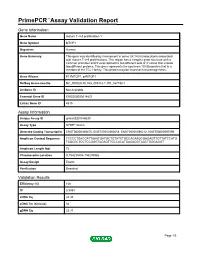
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name mature T-cell proliferation 1 Gene Symbol MTCP1 Organism Human Gene Summary This gene was identified by involvement in some t(X;14) translocations associated with mature T-cell proliferations. This region has a complex gene structure with a common promoter and 5' exon spliced to two different sets of 3' exons that encode two different proteins. This gene represents the upstream 13 kDa protein that is a member of the TCL1 family. This protein may be involved in leukemogenesis. Gene Aliases P13MTCP1, p8MTCP1 RefSeq Accession No. NC_000023.10, NG_005114.1, NT_167198.1 UniGene ID Not Available Ensembl Gene ID ENSG00000214827 Entrez Gene ID 4515 Assay Information Unique Assay ID qHsaCED0048630 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000369476, ENST00000362018, ENST00000596212, ENST00000597096 Amplicon Context Sequence TCCCCTGACCATTAAATGATGCTGTATCTGCCACAAGCGAGAGTTGTTATCCATG TAGCGCTCCTCCGGGTAGAGTTGCCACATGAGAGGTAGCTGGGAGGT Amplicon Length (bp) 72 Chromosome Location X:154293804-154293986 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 106 R2 0.9994 cDNA Cq 24.34 cDNA Tm (Celsius) 82 gDNA Cq 25.37 Page 1/5 PrimePCR™Assay Validation Report Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report MTCP1, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard -

Structural Studies of the Complex Between Akt-In and the Akt2-PH Domain Suggest That the Peptide Acts As an Allosteric Inhibitor of the Akt Kinase
The Open Spectroscopy Journal, 2009, 3, 65-76 65 Open Access Structural Studies of the Complex Between Akt-in and the Akt2-PH Domain Suggest that the Peptide Acts as an Allosteric Inhibitor of the Akt Kinase Virginie Ropars1,2,3, Philippe Barthe1,2,3, Chi-Shien Wang4, Wenlung Chen4, Der-Lii M. Tzou4,5, Anne Descours6, Loïc Martin6, Masayuki Noguchi7, Daniel Auguin1,2,3,,¶ and Christian Roumestand*,1,2,3 1CNRS UMR 5048, Centre de Biochimie Structurale, Montpellier, France 2INSERM U554, Montpellier, France 3Université Montpellier I et II, Montpellier, France 4Department of Applied Chemistry, National Chiayi University, Chiayi 60004, Taiwan, ROC 5Institute of Chemistry, Academia Sinica, Nankang, Taipei 11529, Taiwan, ROC 6CEA, iBiTecs, Service d’Ingénierie Moléculaire des Protéines, 91191 Gif sur Yvette, France 7Division of Cancer Biology, Institute for Genetic Medicine, Hokkaido University, N15 W7, Kita-ku, Sapporo 060-0815, Japan Abstract: Serine/threonine kinase Akt plays a central role in the regulation of cell survival and proliferation. Hence, the search for Akt specific inhibitors constitutes an attractive strategy for anticancer therapy. We have previously demonstrated that the proto-oncogene TCL1 coactivates Akt upon binding to its Plekstrin Homology Domain, and we proposed a model for the structure of the complex TCL1:Akt2-PHD. This model led to the rational design of Akt-in, a peptide inhibitor spanning the A ß-strand of human TCL1 that binds Akt2 PH domain and inhibits the kinase activation. In the present report, we used NMR spectroscopy to determine the 3D structure of the peptide free in solution and bound to Akt2-PHD. -

Association of Gene Ontology Categories with Decay Rate for Hepg2 Experiments These Tables Show Details for All Gene Ontology Categories
Supplementary Table 1: Association of Gene Ontology Categories with Decay Rate for HepG2 Experiments These tables show details for all Gene Ontology categories. Inferences for manual classification scheme shown at the bottom. Those categories used in Figure 1A are highlighted in bold. Standard Deviations are shown in parentheses. P-values less than 1E-20 are indicated with a "0". Rate r (hour^-1) Half-life < 2hr. Decay % GO Number Category Name Probe Sets Group Non-Group Distribution p-value In-Group Non-Group Representation p-value GO:0006350 transcription 1523 0.221 (0.009) 0.127 (0.002) FASTER 0 13.1 (0.4) 4.5 (0.1) OVER 0 GO:0006351 transcription, DNA-dependent 1498 0.220 (0.009) 0.127 (0.002) FASTER 0 13.0 (0.4) 4.5 (0.1) OVER 0 GO:0006355 regulation of transcription, DNA-dependent 1163 0.230 (0.011) 0.128 (0.002) FASTER 5.00E-21 14.2 (0.5) 4.6 (0.1) OVER 0 GO:0006366 transcription from Pol II promoter 845 0.225 (0.012) 0.130 (0.002) FASTER 1.88E-14 13.0 (0.5) 4.8 (0.1) OVER 0 GO:0006139 nucleobase, nucleoside, nucleotide and nucleic acid metabolism3004 0.173 (0.006) 0.127 (0.002) FASTER 1.28E-12 8.4 (0.2) 4.5 (0.1) OVER 0 GO:0006357 regulation of transcription from Pol II promoter 487 0.231 (0.016) 0.132 (0.002) FASTER 6.05E-10 13.5 (0.6) 4.9 (0.1) OVER 0 GO:0008283 cell proliferation 625 0.189 (0.014) 0.132 (0.002) FASTER 1.95E-05 10.1 (0.6) 5.0 (0.1) OVER 1.50E-20 GO:0006513 monoubiquitination 36 0.305 (0.049) 0.134 (0.002) FASTER 2.69E-04 25.4 (4.4) 5.1 (0.1) OVER 2.04E-06 GO:0007050 cell cycle arrest 57 0.311 (0.054) 0.133 (0.002) -

CASP)-Round V
PROTEINS: Structure, Function, and Genetics 53:334–339 (2003) Critical Assessment of Methods of Protein Structure Prediction (CASP)-Round V John Moult,1 Krzysztof Fidelis,2 Adam Zemla,2 and Tim Hubbard3 1Center for Advanced Research in Biotechnology, University of Maryland Biotechnology Institute, Rockville, Maryland 2Biology and Biotechnology Research Program, Lawrence Livermore National Laboratory, Livermore, California 3Sanger Institute, Wellcome Trust Genome Campus, Cambridgeshire, United Kingdom ABSTRACT This article provides an introduc- The role and importance of automated servers in the tion to the special issue of the journal Proteins structure prediction field continue to grow. Another main dedicated to the fifth CASP experiment to assess the section of the issue deals with this topic. The first of these state of the art in protein structure prediction. The articles describes the CAFASP3 experiment. The goal of article describes the conduct, the categories of pre- CAFASP is to assess the state of the art in automatic diction, and the evaluation and assessment proce- methods of structure prediction.16 Whereas CASP allows dures of the experiment. A brief summary of progress any combination of computational and human methods, over the five CASP experiments is provided. Related CAFASP captures predictions directly from fully auto- developments in the field are also described. Proteins matic servers. CAFASP makes use of the CASP target 2003;53:334–339. © 2003 Wiley-Liss, Inc. distribution and prediction collection infrastructure, but is otherwise independent. The results of the CAFASP3 experi- Key words: protein structure prediction; communi- ment were also evaluated by the CASP assessors, provid- tywide experiment; CASP ing a comparison of fully automatic and hybrid methods. -

HMMER User's Guide
HMMER User’s Guide Biological sequence analysis using profile hidden Markov models http://hmmer.org/ Version 3.0rc1; February 2010 Sean R. Eddy for the HMMER Development Team Janelia Farm Research Campus 19700 Helix Drive Ashburn VA 20147 USA http://eddylab.org/ Copyright (C) 2010 Howard Hughes Medical Institute. Permission is granted to make and distribute verbatim copies of this manual provided the copyright notice and this permission notice are retained on all copies. HMMER is licensed and freely distributed under the GNU General Public License version 3 (GPLv3). For a copy of the License, see http://www.gnu.org/licenses/. HMMER is a trademark of the Howard Hughes Medical Institute. 1 Contents 1 Introduction 5 How to avoid reading this manual . 5 How to avoid using this software (links to similar software) . 5 What profile HMMs are . 5 Applications of profile HMMs . 6 Design goals of HMMER3 . 7 What’s still missing in HMMER3 . 8 How to learn more about profile HMMs . 9 2 Installation 10 Quick installation instructions . 10 System requirements . 10 Multithreaded parallelization for multicores is the default . 11 MPI parallelization for clusters is optional . 11 Using build directories . 12 Makefile targets . 12 3 Tutorial 13 The programs in HMMER . 13 Files used in the tutorial . 13 Searching a sequence database with a single profile HMM . 14 Step 1: build a profile HMM with hmmbuild . 14 Step 2: search the sequence database with hmmsearch . 16 Searching a profile HMM database with a query sequence . 22 Step 1: create an HMM database flatfile . 22 Step 2: compress and index the flatfile with hmmpress . -

Regulation of the Akt Kinase by Interacting Proteins
Oncogene (2005) 24, 7401–7409 & 2005 Nature Publishing Group All rights reserved 0950-9232/05 $30.00 www.nature.com/onc Regulation of the Akt kinase by interacting proteins Keyong Du1 and Philip N Tsichlis*,1 1Molecular Oncology Research Institute, Tufts-New England Medical Center, Boston, MA 02111, USA Ten years ago, it was observed that the Akt kinase revolutionized our understanding of cell function at is activated by phosphorylation via a phosphoinositide the molecular level.These technologies also allowed 3-kinase (PI-3K)-dependent process. This discovery gene- the identification of a large array of protein–protein rated enormous interest because it provided a link between interaction motifs that facilitate our understanding of PI-3K, an enzyme known to play a critical role in cellular signal transduction by providing clues on the potential physiology, and its downstream targets. Subsequently, it function of novel signaling proteins (Barnouin, 2004; was shown that the activity of the core components of the Miller and Stagljar, 2004). ‘PI-3K/Akt pathway’ is modulated by a complex network Akt1, also known as protein kinase Ba (PKBa) of regulatory proteins and pathways. Some of the Akt- (Bellacosa et al., 1991; Coffer and Woodgett, 1991; binding partners modulate its activation by external Jones et al., 1991), is the founding member of a protein signals by interacting with different domains of the Akt kinase family composed of three members, Akt1, Akt2, protein. This review focuses on the Akt interacting and Akt3.Akt family members regulate a diverse proteins and the mechanisms by which they regulate Akt array of cellular functions, including apoptosis, cellular activation. -

Benchmarking the POEM@HOME Network for Protein Structure
3rd International Workshop on Science Gateways for Life Sciences (IWSG 2011), 8-10 JUNE 2011 Benchmarking the POEM@HOME Network for Protein Structure Prediction Timo Strunk1, Priya Anand1, Martin Brieg2, Moritz Wolf1, Konstantin Klenin2, Irene Meliciani1, Frank Tristram1, Ivan Kondov2 and Wolfgang Wenzel1,* 1Institute of Nanotechnology, Karlsruhe Institute of Technology, PO Box 3640, 76021 Karlsruhe, Germany. 2Steinbuch Centre for Computing, Karlsruhe Institute of Technology, PO Box 3640, 76021 Karlsruhe, Germany ABSTRACT forcefields (Fitzgerald, et al., 2007). Knowledge-based potentials, in contrast, perform very well in differentiating native from non- Motivation: Structure based methods for drug design offer great native protein structures (Wang, et al., 2004; Zhou, et al., 2007; potential for in-silico discovery of novel drugs but require accurate Zhou, et al., 2006) and have recently made inroads into the area of models of the target protein. Because many proteins, in particular protein folding. Physics-based models retain the appeal of high transmembrane proteins, are difficult to characterize experimentally, transferability, but the present lack of truly transferable potentials methods of protein structure prediction are required to close the gap calls for the development of novel forcefields for protein structure prediction and modeling (Schug, et al., 2006; Verma, et al., 2007; between sequence and structure information. Established methods Verma and Wenzel, 2009). for protein structure prediction work well only for targets of high We have earlier reported the rational development of transferable homology to known proteins, while biophysics based simulation free energy forcefields PFF01/02 (Schug, et al., 2005; Verma and methods are restricted to small systems and require enormous Wenzel, 2009) that correctly predict the native conformation of computational resources. -

Methods for the Refinement of Protein Structure 3D Models
International Journal of Molecular Sciences Review Methods for the Refinement of Protein Structure 3D Models Recep Adiyaman and Liam James McGuffin * School of Biological Sciences, University of Reading, Reading RG6 6AS, UK; [email protected] * Correspondence: l.j.mcguffi[email protected]; Tel.: +44-0-118-378-6332 Received: 2 April 2019; Accepted: 7 May 2019; Published: 1 May 2019 Abstract: The refinement of predicted 3D protein models is crucial in bringing them closer towards experimental accuracy for further computational studies. Refinement approaches can be divided into two main stages: The sampling and scoring stages. Sampling strategies, such as the popular Molecular Dynamics (MD)-based protocols, aim to generate improved 3D models. However, generating 3D models that are closer to the native structure than the initial model remains challenging, as structural deviations from the native basin can be encountered due to force-field inaccuracies. Therefore, different restraint strategies have been applied in order to avoid deviations away from the native structure. For example, the accurate prediction of local errors and/or contacts in the initial models can be used to guide restraints. MD-based protocols, using physics-based force fields and smart restraints, have made significant progress towards a more consistent refinement of 3D models. The scoring stage, including energy functions and Model Quality Assessment Programs (MQAPs) are also used to discriminate near-native conformations from non-native conformations. Nevertheless, there are often very small differences among generated 3D models in refinement pipelines, which makes model discrimination and selection problematic. For this reason, the identification of the most native-like conformations remains a major challenge. -

Validation and Annotation of Virus Sequence Submissions to Genbank Alejandro A
Schäffer et al. BMC Bioinformatics (2020) 21:211 https://doi.org/10.1186/s12859-020-3537-3 SOFTWARE Open Access VADR: validation and annotation of virus sequence submissions to GenBank Alejandro A. Schäffer1,2, Eneida L. Hatcher2, Linda Yankie2, Lara Shonkwiler2,3,J.RodneyBrister2, Ilene Karsch-Mizrachi2 and Eric P. Nawrocki2* *Correspondence: [email protected] Abstract 2National Center for Biotechnology Background: GenBank contains over 3 million viral sequences. The National Center Information, National Library of for Biotechnology Information (NCBI) previously made available a tool for validating Medicine, National Institutes of Health, Bethesda, MD, 20894 USA and annotating influenza virus sequences that is used to check submissions to Full list of author information is GenBank. Before this project, there was no analogous tool in use for non-influenza viral available at the end of the article sequence submissions. Results: We developed a system called VADR (Viral Annotation DefineR) that validates and annotates viral sequences in GenBank submissions. The annotation system is based on the analysis of the input nucleotide sequence using models built from curated RefSeqs. Hidden Markov models are used to classify sequences by determining the RefSeq they are most similar to, and feature annotation from the RefSeq is mapped based on a nucleotide alignment of the full sequence to a covariance model. Predicted proteins encoded by the sequence are validated with nucleotide-to-protein alignments using BLAST. The system identifies 43 types of “alerts” that (unlike the previous BLAST-based system) provide deterministic and rigorous feedback to researchers who submit sequences with unexpected characteristics. VADR has been integrated into GenBank’s submission processing pipeline allowing for viral submissions passing all tests to be accepted and annotated automatically, without the need for any human (GenBank indexer) intervention. -

Advances in Rosetta Protein Structure Prediction on Massively Parallel Systems
UC San Diego UC San Diego Previously Published Works Title Advances in Rosetta protein structure prediction on massively parallel systems Permalink https://escholarship.org/uc/item/87g6q6bw Journal IBM Journal of Research and Development, 52(1) ISSN 0018-8646 Authors Raman, S. Baker, D. Qian, B. et al. Publication Date 2008 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Advances in Rosetta protein S. Raman B. Qian structure prediction on D. Baker massively parallel systems R. C. Walker One of the key challenges in computational biology is prediction of three-dimensional protein structures from amino-acid sequences. For most proteins, the ‘‘native state’’ lies at the bottom of a free- energy landscape. Protein structure prediction involves varying the degrees of freedom of the protein in a constrained manner until it approaches its native state. In the Rosetta protein structure prediction protocols, a large number of independent folding trajectories are simulated, and several lowest-energy results are likely to be close to the native state. The availability of hundred-teraflop, and shortly, petaflop, computing resources is revolutionizing the approaches available for protein structure prediction. Here, we discuss issues involved in utilizing such machines efficiently with the Rosetta code, including an overview of recent results of the Critical Assessment of Techniques for Protein Structure Prediction 7 (CASP7) in which the computationally demanding structure-refinement process was run on 16 racks of the IBM Blue Gene/Le system at the IBM T. J. Watson Research Center. We highlight recent advances in high-performance computing and discuss future development paths that make use of the next-generation petascale (.1012 floating-point operations per second) machines. -

Gathering Them in to the Fold
© 1996 Nature Publishing Group http://www.nature.com/nsmb • comment tion trials, preferring the shorter The first was held in 19947 (see Gathering sequences which are members of a http://iris4.carb.nist.gov/); the sec sequence family: shorter sequences ond will run throughout 19968 were felt to be easier to predict, and ( CASP2: second meeting for the them in to methods such as secondary struc critical assessment of methods of ture prediction are significantly protein structure prediction, Asilo the fold more accurate when based on a mar, December 1996; URL: multiple sequence alignment than http://iris4.carb.nist.gov/casp2/ or A definition of the state of the art in on a single sequence: one or two http://www.mrc-cpe.cam.ac.uk/casp the prediction of protein folding homologous sequences whisper 21). pattern from amino acid sequence about their three-dimensional What do the 'hard' results of the was the object of the course/work structure; a full multiple alignment past predication competition and shop "Frontiers of protein structure shouts out loud. the 'soft' results of the feelings of prediction" held at the Istituto di Program systems used included the workshop partiCipants say 1 Ricerche di Biologia Molecolare fold recognition methods by Sippl , about the possibilities of meaning (IRBM), outside Rome, on 8-17 Jones2, Barton (unpublished) and ful prediction? Certainly it is worth October 1995 (organized by T. Rost 3; secondary structure predic trying these methods on your target Hubbard & A. Tramontano; see tion by Rost and Sander\ analysis sequence-if you find nothing http://www.mrc-cpe.cam.ac.uk/irbm of correlated mutations by Goebel, quickly you can stop and return to course951). -

Improving the Performance of Rosetta Using Multiple Sequence Alignment
PROTEINS:Structure,Function,andGenetics43:1–11(2001) ImprovingthePerformanceofRosettaUsingMultiple SequenceAlignmentInformationandGlobalMeasures ofHydrophobicCoreFormation RichardBonneau,1 CharlieE.M.Strauss,2 andDavidBaker1* 1DepartmentofBiochemistry,Box357350,UniversityofWashington,Seattle,Washington 2BiosciencesDivision,LosAlamosNationalLaboratory,LosAlamos,NewMexico ABSTRACT Thisstudyexplorestheuseofmul- alwayssharethesamefold.8–11 Eachsequencealignment tiplesequencealignment(MSA)informationand canthereforebethoughtofasrepresentingasinglefold, globalmeasuresofhydrophobiccoreformationfor witheachpositionintheMSArepresentingthepreference improvingtheRosettaabinitioproteinstructure fordifferentaminoacidsandgapsatthecorresponding predictionmethod.Themosteffectiveuseofthe positioninthefold.BadretdinovandFinkelsteinand MSAinformationisachievedbycarryingoutinde- colleaguesusedthetheoreticalframeworkoftherandom pendentfoldingsimulationsforasubsetofthe energymodeltoarguethatthedecoydiscrimination homologoussequencesintheMSAandthenidentify- problemisnotcurrentlypossibleunlesstheinformationin ingthefreeenergyminimacommontoallfolded theMSAisusedtosmooththeenergylandscape.12,13 sequencesviasimultaneousclusteringoftheinde- Usingathree-dimensional(3D)cubiclatticemodelfora pendentfoldingruns.Globalmeasuresofhydropho- polypeptide,theseinvestigatorsdemonstratedthataverag- biccoreformation,usingellipsoidalratherthan ingascoringfunctioncontainingrandomerrorsover sphericalrepresentationsofthehydrophobiccore, severalhomologoussequencesallowedthecorrectfoldto