Deeply Conserved Chordate Non-Coding Sequences Preserve Genome Synteny but Do Not Drive Gene Duplicate Retention
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Canestro 06Evodev Retinoic Acid Machinery in Non-Chordates.Pdf
EVOLUTION & DEVELOPMENT 8:5, 394–406 (2006) Is retinoic acid genetic machinery a chordate innovation? Cristian Can˜estro,a John H. Postlethwait,a Roser Gonza`lez-Duarte,b and Ricard Albalatb,Ã aInstitute of Neuroscience, University of Oregon, Eugene, OR 97403, USA bDepartament de Gene`tica, Universitat de Barcelona, Av. Diagonal 645, 08028 Barcelona, Spain ÃAuthor for correspondence (email: [email protected]) SUMMARY Development of many chordate features and showed for the first time that RA genetic machineryF depends on retinoic acid (RA). Because the action of RA that is Aldh1a, Cyp26, and Rar orthologsFis present in during development seems to be restricted to chordates, it had nonchordate deuterostomes. This finding implies that RA been previously proposed that the ‘‘invention’’ of RA genetic genetic machinery was already present during early machinery, including RA-binding nuclear hormone receptors deuterostome evolution, and therefore, is not a chordate (Rars), and the RA-synthesizing and RA-degrading enzymes innovation. This new evolutionary viewpoint argues against Aldh1a (Raldh) and Cyp26, respectively, was an important the hypothesis that the acquisition of gene families under- step for the origin of developmental mechanisms leading lying RA metabolism and signaling was a key event for to the chordate body plan. We tested this hypothesis the origin of chordates. We propose a new hypothesis in by conducting an exhaustive survey of the RA machinery which lineage-specific duplication and loss of RA machinery in genomic databases for twelve deuterostomes. We genes could be related to the morphological radiation of reconstructed the evolution of these genes in deuterostomes deuterostomes. INTRODUCTION which appears to be the sister group of chordates (Cameron et al. -

Takifugu Niphobles
Joseph Fratello Marine Biology Professor Tudge 10/16/17 Takifugu niphobles Introduction: The Takifugu niphobles or the Grass Puffer is a small fish that resides in the shallow waters of the Northwest Pacific Ocean. The scientific name of the fish comes from the japanese words of taki meaning waterfall and fugu meaning venomous fish (Torres, Armi G., et al). The Takifugu niphobles is part of the family Tetraodontidae which encompasses all puffer fish and are known for their ability to inflate like a balloon. The fish do this by quickly sucking water into their stomachs causing them to inflate and causing the flat lying spines which cover their bodies to become erect. Their diets consist of a wide array of small crustaceans and mollusks (Practical Fishkeeping, 2010). Takifugu niphobles are one of the two most common fish in the Northwest Pacific Ocean and are often accidently caught by fishermen who employ the bottom longline technique (Shao K, et al., 2014). The sale of these fish, including other puffers, are banned in japanese markets due to their highly toxic nature. Yet, puffer fish are considered a japanese delicacy despite the fact that a wrong cut of meat can kill a fully grown man. Upwards of thirty to fifty people are affected by the toxin every year and chefs must undergo two years of training before they can legally sell the fish (Dan Bloom 2015). These fish have a very unique means of reproduction, in which they swim towards the shore and lay their eggs on the beach. The fish then, with the help of the waves, beach themselves and fertilize these eggs. -

Acceptability Evaluation by Vietnamese About Non-Toxic Cultured Pufferfish in Comparison with Grouper and Mackerel
Asian Journal of Dietetics 2019 ORIGINAL Acceptability Evaluation by Vietnamese about Non-toxic Cultured Pufferfish in Comparison with Grouper and Mackerel Linh Vu Thuy1, Tuyen Le Danh3, Hien Vu Thi Thu3, Bat Nguyen Khac4, Ngoc Vuong Thi Ho3, Nghia Nguyen Viet4, Hien Bui Thi Thu4, Fumio Shimura1, Sumiko Kamoshita1, Yoshinari Ito2, Shigeru Yamamoto1 1 International Nutrition, Graduate School of Human Life Sciences, Jumonji University,Saitama 352-8510, Japan 2Mitsui Marine Products Inc, Miyazaki 889-0511, Japan 3 Vietnam National Institute of Nutrition, Hanoi, Vietnam 4 Vietnam Marine Fisheries Research Institute, Hai Phong, Vietnam (Recived June 4,2019) ABSTRACT Background and purpose. World-wide there are few countries in which pufferfish (fugu) is eaten as in Japan. In Vietnam, pufferfish has been banned since it’s toxic ocurred due to lack of knowledge about distinguish non-toxic species. Consequently, there is a huge amount of pufferfish inhabiting in Vietnamese water, but whenever accidentally catching it, we have to throw or use as fertilizer. To develop culinary culture of non-toxic pufferfish in Vietnam, it is very important and essential to recognize safe species and culture them in good condition, then apply proper method to process that become safety foods. In order to initial setting up for such purpose, we carried out a sensory study in Vietnamese to test whether they can accept foods made from fugu by method of Japanese. Methods. We compared the sensory reaction to Japanese cultured Takifugu rubripes and fish from Vietnamese waters: grouper and mackerel. The 107 panelists were Vietnamese volunteers working in the field of nutrition, employees of marine companies, or government officials who could influence the relevant laws. -

Takif Ugu O Bscurus
MARINE ECOLOGY PROGRESS SERIES Published November 14 Mar Ecol Prog Ser Effects of pentachlorophenol (PCP) on the oxygen consumption rate of the river puffer fish Takif ugu o bscurus 'Marine Biotechnology Lab., and 'Chemical Oceanography Division. Korea Ocean Research & Development Institute, Ansan. PO Box 29. Seoul 425-600. Korea ABSTRACT: Laboratory bioassays were conducted to determine the effects of pentachlorophenol (PCP) on the oxygen consumption of the river puffer fish Takifugu ohscnrus. Oxygen consu.mption of 6 to 9 mo old puffer fish was measured with an automatic intermittent-flow-respirometer (AIFR).Oxygen consumption rate was s~gnificantlyincreased by exposing fish to concentrations of 50, 100, 200 and 500 ppb PCP Follow~ngexposure to 50 or 100 ppb PCP, the instantaneous rate of oxygen consumption was considerably increased. T obscurus exposed to 200 ppb PCP, however, exhibited a breakdown in the biorhythm of oxygen consumption presumably due to a strong physiological stress caused by the higher PCP concentrations. River puffer fish exposed to 500 ppb PCP died in 10 h. KEY WORDS. PCP . Oxygen consumption . River puffer f~sh Tak~fuguohscurus INTRODUCTION one way to decipher the bio-physico-chemical linkage would be to test the effects of a variety of chemicals on Pentachlorophenol (PCP) is a widely used biocide organism biorhythms, i.e. how they alter their periods (Cirelli 1978), even though it is known to be highly or phase. Despite the well-documented role of PCP toxic to most living organisms. Samis et al. (1993) in biochemical responses (Coglianese & Jerry 1982, reported that growth rate of bluegills Lepomis macro- Yousrl& Hanke 1985, Castren & Oikari 1987) and eco- chirus was significantly reduced at a PCP concentra- logical processes (Whitney et al. -

Phylogenetic Analysis of the Tenascin Gene Family: Evidence of Origin
BMC Evolutionary Biology BioMed Central Research article Open Access Phylogenetic analysis of the tenascin gene family: evidence of origin early in the chordate lineage RP Tucker*1, K Drabikowski*2,4, JF Hess1, J Ferralli2, R Chiquet-Ehrismann2 and JC Adams3 Address: 1Department of Cell Biology and Human Anatomy, University of California at Davis, Davis, CA 95616, USA, 2Friedrich Miescher Institute, Novartis Research Foundation, Basel, Switzerland, 3Dept. of Cell Biology, Lerner Research Institute and Dept. of Molecular Medicine, Cleveland Clinic Lerner College of Medicine, Cleveland Clinic Foundation, Cleveland, OH 44118, USA and 4Institute of Biology 3, University of Freiburg, Freiburg, Germany Email: RP Tucker* - [email protected]; K Drabikowski* - [email protected]; JF Hess - [email protected]; J Ferralli - [email protected]; R Chiquet-Ehrismann - [email protected]; JC Adams - [email protected] * Corresponding authors Published: 07 August 2006 Received: 24 February 2006 Accepted: 07 August 2006 BMC Evolutionary Biology 2006, 6:60 doi:10.1186/1471-2148-6-60 This article is available from: http://www.biomedcentral.com/1471-2148/6/60 © 2006 Tucker et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Tenascins are a family of glycoproteins -

Plasma Protein Binding of Tetrodotoxin in the Marine Puffer Fish Takifugu
Toxicon 55 (2010) 415–420 Contents lists available at ScienceDirect Toxicon journal homepage: www.elsevier.com/locate/toxicon Plasma protein binding of tetrodotoxin in the marine puffer fish Takifugu rubripes Takuya Matsumoto a, Daisuke Tanuma a, Kazuma Tsutsumi a, Joong-Kyun Jeon b, Shoichiro Ishizaki a, Yuji Nagashima a,* a Department of Food Science and Technology, Tokyo University of Marine Science and Technology, Konan 4-5-7, Minato, Tokyo 108-8477, Japan b Division of Marine Bioscience and Technology, Kangnung-Wonju National University, Jibyeon, Gangneung 210-702, Republic of Korea article info abstract Article history: To elucidate the involvement of plasma protein binding in the disposition of tetrodotoxin Received 15 June 2009 (TTX) in puffer fish, we used equilibrium dialysis to measure protein binding of TTX in the Received in revised form plasma of the marine puffer fish Takifugu rubripes and the non-toxic greenling Hexagrammos 31 August 2009 otakii, and in solutions of bovine serum albumin (BSA) and bovine alpha-1-acid glycoprotein Accepted 15 September 2009 (AGP). TTX (100–1000 mg/mL) bound to protein in T. rubripes plasma with low affinity in Available online 22 September 2009 a non-saturable manner. The amount of bound TTX increased linearly with the TTX concentration, reaching 3.92 Æ 0.42 mg TTX/mg protein at 1000 mg TTX/mL. Approximately Keywords: Tetrodotoxin 80% of the TTX in the plasma of T. rubripes was unbound in the concentration range of TTX Plasma protein binding examined, indicating that TTX exists predominantly in the unbound form in the circulating Equilibrium dialysis blood of T. -

SAIA List of Ecologically Unsustainable Species
SAIA List of Ecologically Unsustainable Species Note The aquarium fishery in Southeast Asia contributes to the destruction of coral reefs. Although illegal, the use of cyanide to stun fish is still widespread, especially for species that seek shelter between coral branches, in holes, and among rocks (like damsels or gobies), but also those occurring at greater depths (e.g., dwarf angels, some anthias) or the ones fetching high prices (like angelfish or surgeonfish). While ideally the dosage is only intended to stun the targeted fish, it is often sufficient to kill the non-targeted invertebrates building the reef. As such, is a destructive fishing method, banned by regulation in Indonesia and the Philippines. Fish caught with cyanide are a product of illegal fishing. According to EU Regulation, the import of products from illegal, unreported, and unregulated (IUU) fishing is prohibited.* Similarly, the Lacey Act, a conservation law in the United States, prohibits trade in wildlife, fish, and plants that have been illegally taken, possessed, transported, or sold. However, enforcing these laws is difficult because there is insufficient control in both the countries of origin and in the markets. Therefore, the likelihood of purchasing a product from illegal fishing is real. Ask your dealer about the origin of the offered animals and insist on sustainable fishing methods! Inadequate or deficient fishery management is another, often underestimated, problem of aquarium fisheries in South East Asia. Many fish come from unreported and unregulated fisheries. For most coral fish species, but also invertebrates, no data exist. The status of local populations and catch volumes are thus unknown. -
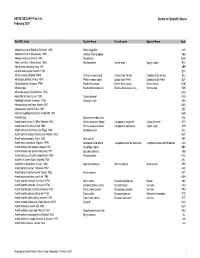
ASFIS ISSCAAP Fish List February 2007 Sorted on Scientific Name
ASFIS ISSCAAP Fish List Sorted on Scientific Name February 2007 Scientific name English Name French name Spanish Name Code Abalistes stellaris (Bloch & Schneider 1801) Starry triggerfish AJS Abbottina rivularis (Basilewsky 1855) Chinese false gudgeon ABB Ablabys binotatus (Peters 1855) Redskinfish ABW Ablennes hians (Valenciennes 1846) Flat needlefish Orphie plate Agujón sable BAF Aborichthys elongatus Hora 1921 ABE Abralia andamanika Goodrich 1898 BLK Abralia veranyi (Rüppell 1844) Verany's enope squid Encornet de Verany Enoploluria de Verany BLJ Abraliopsis pfefferi (Verany 1837) Pfeffer's enope squid Encornet de Pfeffer Enoploluria de Pfeffer BJF Abramis brama (Linnaeus 1758) Freshwater bream Brème d'eau douce Brema común FBM Abramis spp Freshwater breams nei Brèmes d'eau douce nca Bremas nep FBR Abramites eques (Steindachner 1878) ABQ Abudefduf luridus (Cuvier 1830) Canary damsel AUU Abudefduf saxatilis (Linnaeus 1758) Sergeant-major ABU Abyssobrotula galatheae Nielsen 1977 OAG Abyssocottus elochini Taliev 1955 AEZ Abythites lepidogenys (Smith & Radcliffe 1913) AHD Acanella spp Branched bamboo coral KQL Acanthacaris caeca (A. Milne Edwards 1881) Atlantic deep-sea lobster Langoustine arganelle Cigala de fondo NTK Acanthacaris tenuimana Bate 1888 Prickly deep-sea lobster Langoustine spinuleuse Cigala raspa NHI Acanthalburnus microlepis (De Filippi 1861) Blackbrow bleak AHL Acanthaphritis barbata (Okamura & Kishida 1963) NHT Acantharchus pomotis (Baird 1855) Mud sunfish AKP Acanthaxius caespitosa (Squires 1979) Deepwater mud lobster Langouste -

Record and Distribution of Puffer Fish, Takifugu Oblongus (Bloch, 1786) (Actinopterygii: Tetraodontidnae) from Tapi River, Gujarat
Egyptian Journal of Aquatic Biology & Fisheries Zoology Department, Faculty of Science, Ain Shams University, Cairo, Egypt. ISSN 1110 - 6131 Vol. 22(5): 401- 404 (2018) ejabf.journals.ekb.eg Record and distribution of puffer fish, Takifugu oblongus (Bloch, 1786) (Actinopterygii: Tetraodontidnae) from Tapi River, Gujarat. Thakkar Nevya1, Sarma Kangkan Jyoti1, Tatu Ketan2, Kamboj Ravi D.2 and Mankodi Pradeep1* 1- Division of Freshwater and Marine Biology, Department of Zoology, Faculty of Science, The Maharaja Sayajirao Univ. Baroda, Vadodara – 390002, Gujarat, India. 2- Gujarat Ecological Education and Research (GEER) Foundation, Indroda Nature Park, Sector – 7, Gandhinagar – 382007, Gujarat, India. *Corresponding author: [email protected] ARTICLE INFO ABSTRACT Article History: Takifugu oblongus (Bloch, 1786) (Actinopterygii: Received: July, 24, 2018 Tetraodontidae), is a puffer fish found in shallow coastal waters has a Accepted: Dec. 15, 2018 native distribution in entire Indo-West Pacific region (Froese and Online: Dec. 28, 2018 Pauly, 2014). This species is observed in shallow coastal ecosystems _______________ such as estuaries, mangroves, coral reefs and sand and gravel Keywords: sediments, including the soft bottom areas of inshore and offshore Puffer fish regions. Takifugu oblongus is one of the widely distributed species Tetraodontidae among the genus Takigufu but no report or publication has been Distribution made so far from the western coast of India. This paper reports the Tapi River Gujarat. record of the fish from Magdalla along the course of Tapi River, Surat, Gujarat. INTRODUCTION Puffer fishes are oval shaped and possess spines all around their bodies with toxic glands beneath. They have the ability to inflate or expand their bodies with air or water when they feel threatened and are most commonly found in marine or brackish water habitats (Nelson, 1994; Shipp, 2003). -

Genome Duplication in the Teleost Fish Tetraodon Nigroviridis Reveals The
articles Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype Olivier Jaillon1, Jean-Marc Aury1, Fre´de´ric Brunet2, Jean-Louis Petit1, Nicole Stange-Thomann3, Evan Mauceli3, Laurence Bouneau1, Ce´cile Fischer1, Catherine Ozouf-Costaz4, Alain Bernot1, Sophie Nicaud1, David Jaffe3, Sheila Fisher3, Georges Lutfalla5, Carole Dossat1, Be´atrice Segurens1, Corinne Dasilva1, Marcel Salanoubat1, Michael Levy1, Nathalie Boudet1, Sergi Castellano6,Ve´ronique Anthouard1, Claire Jubin1, Vanina Castelli1, Michael Katinka1, Benoıˆt Vacherie1, Christian Bie´mont7, Zineb Skalli1, Laurence Cattolico1, Julie Poulain1, Ve´ronique de Berardinis1, Corinne Cruaud1, Simone Duprat1, Philippe Brottier1, Jean-Pierre Coutanceau4,Je´roˆme Gouzy8, Genis Parra6, Guillaume Lardier1, Charles Chapple6, Kevin J. McKernan9, Paul McEwan9, Stephanie Bosak9, Manolis Kellis3, Jean-Nicolas Volff10, Roderic Guigo´ 6, Michael C. Zody3, Jill Mesirov3, Kerstin Lindblad-Toh3, Bruce Birren3, Chad Nusbaum3, Daniel Kahn8, Marc Robinson-Rechavi2, Vincent Laudet2, Vincent Schachter1, Francis Que´tier1, William Saurin1, Claude Scarpelli1, Patrick Wincker1, Eric S. Lander3,11, Jean Weissenbach1 & Hugues Roest Crollius1* 1UMR 8030 Genoscope, CNRS and Universite´ d’Evry, 2 rue Gaston Cre´mieux, 91057 Evry Cedex, France 2Laboratoire de Biologie Mole´culaire de la Cellule, CNRS UMR 5161, INRA UMR 1237, Ecole Normale Supe´rieure de Lyon, 46 alle´e d’Italie, 69364 Lyon Cedex 07, France 3Broad Institute of MIT and Harvard, 320 Charles Street, -

Retinoic Acid Signaling and the Evolution of Chordates Ferdinand Marlétaz1, Linda Z
Int. J. Biol. Sci. 2006, 2 38 International Journal of Biological Sciences ISSN 1449-2288 www.biolsci.org 2006 2(2):38-47 ©2006 Ivyspring International Publisher. All rights reserved Review Retinoic acid signaling and the evolution of chordates Ferdinand Marlétaz1, Linda Z. Holland2, Vincent Laudet1 and Michael Schubert1 1. Laboratoire de Biologie Moléculaire de la Cellule, CNRS UMR5161/INRA 1237/ENS Lyon, IFR128 BioSciences/Lyon- Gerland, Ecole Normale Supérieure de Lyon, 46 Allée d’Italie, 69364 Lyon Cedex 07, France 2. Marine Biology Research Division, Scripps Institution of Oceanography, University of California San Diego, La Jolla, CA 92093-0202, USA Corresponding address: V. Laudet, Laboratoire de Biologie Moléculaire de la Cellule, CNRS UMR5161/INRA 1237/ENS Lyon, IFR128 BioSciences/Lyon-Gerland, Ecole Normale Supérieure de Lyon, 46 Allée d’Italie, 69364 Lyon Cedex 07, France. E-mail: [email protected] - Tel: ++33 4 72 72 81 90 - Fax: ++33 4 72 72 80 80 Received: 2006.02.13; Accepted: 2006.03.15; Published: 2006.04.10 In chordates, which comprise urochordates, cephalochordates and vertebrates, the vitamin A-derived morphogen retinoic acid (RA) has a pivotal role during development. Altering levels of endogenous RA signaling during early embryology leads to severe malformations, mainly due to incorrect positional codes specifying the embryonic anteroposterior body axis. In this review, we present our current understanding of the RA signaling pathway and its roles during chordate development. In particular, we focus on the conserved roles of RA and its downstream mediators, the Hox genes, in conveying positional patterning information to different embryonic tissues, such as the endoderm and the central nervous system. -

Global Conservation Status of Marine Pufferfishes (Tetraodontiformes: Tetraodontidae) Emilie Stump Old Dominion University
Old Dominion University ODU Digital Commons Biological Sciences Faculty Publications Biological Sciences 4-2018 Global Conservation Status of Marine Pufferfishes (Tetraodontiformes: Tetraodontidae) Emilie Stump Old Dominion University Gina M. Ralph Old Dominion University Mia T. Comeros-Raynal Old Dominion University Keiichi Matsuura Kent E. Carpenter Old Dominion University, [email protected] Follow this and additional works at: https://digitalcommons.odu.edu/biology_fac_pubs Part of the Biodiversity Commons, Ecology and Evolutionary Biology Commons, Environmental Sciences Commons, and the Marine Biology Commons Repository Citation Stump, Emilie; Ralph, Gina M.; Comeros-Raynal, Mia T.; Matsuura, Keiichi; and Carpenter, Kent E., "Global Conservation Status of Marine Pufferfishes (Tetraodontiformes: Tetraodontidae)" (2018). Biological Sciences Faculty Publications. 318. https://digitalcommons.odu.edu/biology_fac_pubs/318 Original Publication Citation Stump, E., Ralph, G. M., Comeros-Raynal, M. T., Matsuura, K., & Carpenter, K. E. (2018). Global conservation status of marine pufferfishes (Tetraodontiformes: Tetraodontidae). Global Ecology and Conservation, 14, e00388. doi:10.1016/j.gecco.2018.e00388 This Article is brought to you for free and open access by the Biological Sciences at ODU Digital Commons. It has been accepted for inclusion in Biological Sciences Faculty Publications by an authorized administrator of ODU Digital Commons. For more information, please contact [email protected]. Global Ecology and Conservation 14 (2018) e00388