Chromosome Breakage Hotspots and Delineation of the Critical Region for the 9P-Deletion Syndrome Laurie A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Abstracts from the 50Th European Society of Human Genetics Conference: Electronic Posters
European Journal of Human Genetics (2019) 26:820–1023 https://doi.org/10.1038/s41431-018-0248-6 ABSTRACT Abstracts from the 50th European Society of Human Genetics Conference: Electronic Posters Copenhagen, Denmark, May 27–30, 2017 Published online: 1 October 2018 © European Society of Human Genetics 2018 The ESHG 2017 marks the 50th Anniversary of the first ESHG Conference which took place in Copenhagen in 1967. Additional information about the event may be found on the conference website: https://2017.eshg.org/ Sponsorship: Publication of this supplement is sponsored by the European Society of Human Genetics. All authors were asked to address any potential bias in their abstract and to declare any competing financial interests. These disclosures are listed at the end of each abstract. Contributions of up to EUR 10 000 (ten thousand euros, or equivalent value in kind) per year per company are considered "modest". Contributions above EUR 10 000 per year are considered "significant". 1234567890();,: 1234567890();,: E-P01 Reproductive Genetics/Prenatal and fetal echocardiography. The molecular karyotyping Genetics revealed a gain in 8p11.22-p23.1 region with a size of 27.2 Mb containing 122 OMIM gene and a loss in 8p23.1- E-P01.02 p23.3 region with a size of 6.8 Mb containing 15 OMIM Prenatal diagnosis in a case of 8p inverted gene. The findings were correlated with 8p inverted dupli- duplication deletion syndrome cation deletion syndrome. Conclusion: Our study empha- sizes the importance of using additional molecular O¨. Kırbıyık, K. M. Erdog˘an, O¨.O¨zer Kaya, B. O¨zyılmaz, cytogenetic methods in clinical follow-up of complex Y. -

Detailed Characterization Of, and Clinical Correlations In, 10 Patients
August 2008 ⅐ Vol. 10 ⅐ No. 8 article Detailed characterization of, and clinical correlations in, 10 patients with distal deletions of chromosome 9p Xueya Hauge, PhD1, Gordana Raca, PhD2, Sara Cooper, MS2, Kristin May, PhD3, Rhonda Spiro, MD4, Margaret Adam, MD2, and Christa Lese Martin, PhD2 Purpose: Deletions of distal 9p are associated with trigonocephaly, mental retardation, dysmorphic facial features, cardiac anomalies, and abnormal genitalia. Previous studies identified a proposed critical region for the consensus phenotype in band 9p23, between 11.8 Mb and 16 Mb from the 9p telomere. Here we report 10 new patients with 9p deletions; 9 patients have clinical features consistent with 9pϪ syndrome, but possess terminal deletions smaller than most reported cases, whereas one individual lacks the 9pϪ phenotype and shows a 140-kb interstitial telomeric deletion inherited from his mother. Methods: We combined fluorescence in situ hybridization and microarray analyses to delineate the size of each deletion. Results: The deletion sizes vary from 800 kb to 12.4 Mb in our patients with clinically relevant phenotypes. Clinical evaluation and comparison showed little difference in physical features with regard to the deletion sizes. Severe speech and language impairment were observed in all patients with clinically relevant phenotypes. Conclusion: The smallest deleted region common to our patients who demonstrate a phenotype consistent with 9pϪ is Ͻ2 Mb of 9pter, which contains six known genes. These genes may contribute to some of the cardinal features of 9p deletion syndrome. Genet Med 2008:10(8): 599–611. Key Words: 9p deletion, FISH, genotype-phenotype correlation, aCGH The 9p deletion syndrome is characterized by trigonoceph- points in 24 patients with visible 9p deletions and breakpoints aly, moderate to severe mental retardation, low-set, mal- at 9p22 or 9p23. -

Sexual Dimorphism in Diverse Metazoans Is Regulated by a Novel Class of Intertwined Zinc Fingers
Downloaded from genesdev.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press Sexual dimorphism in diverse metazoans is regulated by a novel class of intertwined zinc fingers Lingyang Zhu,1,4 Jill Wilken,2 Nelson B. Phillips,3 Umadevi Narendra,3 Ging Chan,1 Stephen M. Stratton,2 Stephen B. Kent,2 and Michael A. Weiss1,3–5 1Center for Molecular Oncology, Departments of Biochemistry & Molecular Biology and Chemistry, The University of Chicago, Chicago, Illinois 60637-5419 USA; 2Gryphon Sciences, South San Francisco, California 94080 USA; 3Department of Biochemistry, Case Western Reserve School of Medicine, Cleveland, Ohio 44106-4935 USA Sex determination is regulated by diverse pathways. Although upstream signals vary, a cysteine-rich DNA-binding domain (the DM motif) is conserved within downstream transcription factors of Drosophila melanogaster (Doublesex) and Caenorhabditis elegans (MAB-3). Vertebrate DM genes have likewise been identified and, remarkably, are associated with human sex reversal (46, XY gonadal dysgenesis). Here we demonstrate that the structure of the Doublesex domain contains a novel zinc module and disordered tail. The module consists of intertwined CCHC and HCCC Zn2+-binding sites; the tail functions as a nascent recognition ␣-helix. Mutations in either Zn2+-binding site or tail can lead to an intersex phenotype. The motif binds in the DNA minor groove without sharp DNA bending. These molecular features, unusual among zinc fingers and zinc modules, underlie the organization of a Drosophila enhancer that integrates sex- and tissue-specific signals. The structure provides a foundation for analysis of DM mutations affecting sexual dimorphism and courtship behavior. -

Early ACCESS Diagnosed Conditions List
Iowa Early ACCESS Diagnosed Conditions Eligibility List List adapted with permission from Early Intervention Colorado To search for a specific word type "Ctrl F" to use the "Find" function. Is this diagnosis automatically eligible for Early Medical Diagnosis Name Other Names for the Diagnosis and Additional Diagnosis Information ACCESS? 6q terminal deletion syndrome Yes Achondrogenesis I Parenti-Fraccaro Yes Achondrogenesis II Langer-Saldino Yes Schinzel Acrocallosal syndrome; ACLS; ACS; Hallux duplication, postaxial polydactyly, and absence of the corpus Acrocallosal syndrome, Schinzel Type callosum Yes Acrodysplasia; Arkless-Graham syndrome; Maroteaux-Malamut syndrome; Nasal hypoplasia-peripheral dysostosis-intellectual disability syndrome; Peripheral dysostosis-nasal hypoplasia-intellectual disability (PNM) Acrodysostosis syndrome Yes ALD; AMN; X-ALD; Addison disease and cerebral sclerosis; Adrenomyeloneuropathy; Siemerling-creutzfeldt disease; Bronze schilder disease; Schilder disease; Melanodermic Leukodystrophy; sudanophilic leukodystrophy; Adrenoleukodystrophy Pelizaeus-Merzbacher disease Yes Agenesis of Corpus Callosum Absence of the corpus callosum; Hypogenesis of the corpus callosum; Dysplastic corpus callosum Yes Agenesis of Corpus Callosum and Chorioretinal Abnormality; Agenesis of Corpus Callosum With Chorioretinitis Abnormality; Agenesis of Corpus Callosum With Infantile Spasms And Ocular Anomalies; Chorioretinal Anomalies Aicardi syndrome with Agenesis Yes Alexander Disease Yes Allan Herndon syndrome Allan-Herndon-Dudley -

2003 Birmingham
11 S1 The Advanced DNA Banking and European Journal of Human Genetics Isolation Technology – IsoCode® IsoCode is optimized lyse Cells IsoCode is available To isolate DNA Standard Card and Stix archive DNA As ID with colour indicator prepare PCR IsoCode can be used high through-put format identification of individuals you want For population screening forensic testing paternity testing • Supplement 11 1 Volume Archiving DNA, cDNA Clones and Vectors The Official Journal of the European Society of Human Genetics Biological samples dried on IsoCode can be archived indefinitely at ambi- ent temperatures. IsoCode is bactericidal, virucidal, and fungicidal. Isolation of DNA without any reagents EUROPEAN HUMAN GENETICS CONFERENCE 2003 IsoCode binds and inactivates proteins and inhibitors – but not the DNA . DNA is released from IsoCode in a simple water elution process that MAY 3 – 6, 2003, BIRMINGHAM, ENGLAND requires neither reagents nor additional costs. PROGRAMME AND ABSTRACTS High quality DNA – easy and cheap DNA eluted from IsoCode is ready for PCR, automated DNA sequencing, STR analysis, mtDNA analysis and other bio-molecular techniques. Use in a lot of fields May 2003 Ideal for drug discovery SNP libraries, epide- miological studies, human identity appli- cations, forensic samples and as back-up copy. EHGC 2003 Booth 940 Volume 11 – Supplement 1 – May 2003 U.S. Pat. #5.939.259, U.S. Pat. 6.168.922 Made under license from Whatman plc. to U.S. Pat. #5.807.527 Schleicher & Schuell BioScience GmbH · Tel. +49-55 61-79 14 63 · Fax +49-55 61-79 15 83 · D-37582 Dassel · Germany · [email protected] Schleicher & Schuell BioScience Inc. -

Association of Distal Deletion of the Short Arm of Chromosome 9 with 46
tems: ys Op l S e a n A ic c g c o l e s o i s B Biological Systems: Open Access Del Rey G et.al., Biol syst Open Access 2015, 4:1 DOI: 10.4172/2329-6577.1000129 ISSN: 2329-6577 Mini Review Open Access Association of Distal Deletion of the Short arm of Chromosome 9 with 46,XY Disorder of Sex Development and Gonadoblastoma Del Rey G1, Venara M1, Papendieck P1,Gruñeiro L1, Tangari A2, Boywitt A1, Casali B1 and Laudicina A3 1Centro de Investigaciones Endocrinológicas, División de Endocrinología, Hospital de Niños, Argentina 2Fundación Hospitalaria, Buenos Aires, Argentina 3Lexel SRL. División in vitro. Buenos Aires, Argentina. *Corresponding author: Graciela del Rey, Centro de Investigaciones, Endocrinológicas, División de, Endocrinología, Hospital de Niños, Gallo 1330 C1425SEFD 29425, Buenos Aires, Argentina, Tel: (54 11) 4963-5931 int 116; Fax: (54 11) 4963-5930, E-mail: [email protected] Received date: Dec 20, 2014; Accepted date: Feb 10, 2015; Published date: Feb17, 2015 Copyright: ©2015. Graciela del Rey, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited Abstract Deletion of the short arm of chromosome 9 is associated with two distinct clinical prototypes. Small telomeric distal 9p deletions have been reported in patients 46,XY with gonadal dysgenesis, this region contains genes required in two copies for normal testis development. Recent studies have narrowed the interval 9p24.3-pter containing the putative autosomal testis-determining gene(s) known as domain DMRT. -
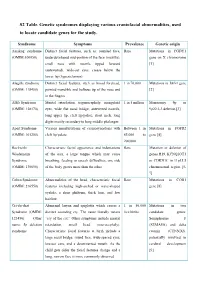
S2 Table. Genetic Syndromes Displaying Various Craniofacial Abnormalities, Used to Locate Candidate Genes for the Study
S2 Table. Genetic syndromes displaying various craniofacial abnormalities, used to locate candidate genes for the study. Syndrome Symptoms Prevalence Genetic origin Aarskog syndrome Distinct facial features, such as: rounded face, Rare Mutations in FGDY1 (OMIM:100050) underdeveloped mid-portion of the face (maxilla), gene on X chromosome small nose with nostrils tipped forward [1] (anteverted), wide-set eyes, crease below the lower lip (hypertelorism) Alagille syndrome Distinct facial features, such as broad forehead, 1 in 70,000 Mutations in JAG1 gene (OMIM: 118450) pointed mandible and bulbous tip of the nose and [2] in the fingers Alfi's Syndrome Mental retardation, trigonocephaly, mongoloid 1 in 5 million Monosomy 9p or (OMIM: 158170) eyes, wide flat nasal bridge, anteverted nostrils, 9p22.2-3 deletion [3] long upper lip, cleft lip/palate, short neck, long digits mostly secondary to long middle phalanges Apert Syndrome Various manifestations of craniosynostosis with Between 1 in Mutations in FGFR2 (OMIM: 101200) cleft lip/palate. 65,000 to gene [4] 200,000 Beckwith- Characteristic facial appearance and indentations Rare Mutation or deletion of Wiedemann of the ears, a large tongue which may cause genes H19, KCNQ1OT1 Syndrome breathing, feeding or speech difficulties, one side or CDKN1C in 11p15.5 (OMIM: 130650) of the body grows more than the other chromosomal region [5- 7] Cohen Syndrome Abnormalities of the head, characteristic facial Rare Mutations in COH1 (OMIM: 216550) features including high-arched or wave-shaped gene [8] eyelids, a short philtrum, thick hair, and low hairline Cri-du-chat Abnormal larynx and epiglottis which causes a 1 in 50,000 Mutations in two Syndrome (OMIM: distinct sounding cry. -

Study of 250 Children with Idiopathic Mental Retardation Reveals Nine Cryptic and Diverse Subtelomeric Chromosome Anomalies
American Journal of Medical Genetics 107:285±293 (2002) Study of 250 Children With Idiopathic Mental Retardation Reveals Nine Cryptic and Diverse Subtelomeric Chromosome Anomalies Elizabeth Baker,1,2 Lyn Hinton,1 David F. Callen,1,3 Meryl Altree,4 Angus Dobbie,4 Helen J. Eyre,1 Grant R. Sutherland,1,3 Elizabeth Thompson,4 Peter Thompson,5 Erica Woollatt,1 and Eric Haan4* 1Centre for Medical Genetics, Department of Cytogenetics and Molecular Genetics, Women's and Children's Hospital, Adelaide, Australia 2Department of Paediatrics, University of Adelaide, Adelaide, Australia 3Department of Molecular Biosciences, University of Adelaide, Adelaide, Australia 4South Australian Clinical Genetics Service, Women's and Children's Hospital, Adelaide, Australia 5University Hospital of Wales, Cardiff, Wales Cryptic subtelomeric chromosome anoma- laboratoryÐthat of choosing the most pro- lies have been recognized as a signi®cant ductive patient base for this useful diagnos- cause of dysmorphology and mental retar- tic test. ß 2001 Wiley-Liss, Inc. dation. To determine whether the clinical cytogenetics laboratory should screen rou- KEY WORDS: idiopathic mental retarda- tinely for these aberrations, we have tested tion; subtelomeric rearran- 250 patients with idiopathic mental retarda- gements; cryptic chromo- tion/developmental delay, either isolated some abnormalities (53) or associated with dysmorphic features and/or malformations in the absence of a recognizable syndrome (197). All had nor- INTRODUCTION mal karyotypes at the 550±850 band level. Subtelomeric anomalies were found in 1/53 Cryptic rearrangements involving the subtelomeric of the ®rst group (1.9%) and 8/197 of the regions of chromosomes have emerged as an important second group (4.1%). -

Mosaic Tetrasomy 9P Case with the Phenotype Mimicking Klinefelter Syndrome and Hyporesponse of Gonadotropin-Stimulated Testosterone Production
Kobe J. Med. Sci., Vol. 53, No. 4, pp. 143-150, 2007 Mosaic Tetrasomy 9p Case with the Phenotype Mimicking Klinefelter Syndrome and Hyporesponse of Gonadotropin-Stimulated Testosterone Production WAKAKO OGINO1, YASUHIRO TAKESHIMA1, ATSUSHI NISHIYAMA1, MARIKO YAGI1, NOBUTOSHI OKA2, and MASAFUMI MATSUO1 1Department of Pediatrics, Kobe University Graduate School of Medicine, 2Department of Urology, Hara Hospital Received 4 January 2007/ Accepted 24 January 2007 Key word: tetrasomy 9p, Klinefelter syndrome, FISH, concealed penis Tetrasomy 9p is a rare clinical syndrome and about 30% of known cases exhibit chromosome mosaicism. The cases with tetrasomy 9p mosaicism have been reported to show the various phenotypes. On the other hand, Klinefelter syndrome is well recognized chromosomal abnormality caused by an additional X chromosome in males (47,XXY), and the characteristic clinical findings include tall stature, immaturity of external genitalia, testicular dysfunction. Here, we report a 10-year-old male with tetrasomy of 9p mosaicism, whose phenotypic feature is mimicking Klinefelter syndrome. He was referred to our hospital for inconspicuous penis. He showed tall height (+2.5 SD). Endocrinological examination revealed the poor testosterone response to human chorionic gonadotropin administration, which indicated the testicular hypofunction, whereas MRI revealed concealed penis as a cause of inconspicuous penis. Because of the phenotype mimicking Klinefelter syndrome, karyotype of his blood lymphocytes was analyzed, and an additional marker chromosome was detected in 6% of the investigated metaphases. Fluorescence in situ hybridization analysis revealed that the marker chromosome was an isochromosome 9p, which resulted in tetrasomy 9p. Chromosome analysis of buccal smear also showed mosaicism for two karyotypes: 5% of cells had the isochromosome of 9p, and the other cells showed normal. -

NIFTYTM Plus
Accredited for compliance with NPAAC Standards and ISO 15189 NIFTYTM Plus - Microdeletion/duplication Syndromes 1 1p36 microdeletion Syndrome 31 Jacobsen Syndrome 2 Van der Woude Syndrome I (VWS) 32 1q41-q42 microdeletion Syndrome 3 12q14 microdeletion Syndrome 33 14q11-q22 deletion Syndrome 4 Feingold Syndrome I 34 Dandy-Walker Syndrome (DWS) 5 Split-Hand/Foot Malformation type 5 (SHFM5) 35 Angelman Syndrome/Prader-Willi Syndrome 6 Glass Syndrome 36 Deafness-infertility Syndrome 7 Holoprosencephaly type 6 (HPE6) 37 15q26 overgrowth Syndrome Microphthalmia type 6 Syndrome, pituitary 8 38 Diaphragmatic hernia, congenital (HCD/DIH1) hypoplasia 9 Rieger Syndrome type 1 (RIEG1) 39 5q21.1-q31.2 deletion Syndrome 10 Cri du Chat (5p deletion) Syndrome 40 16p11.2-p12.2 microdeletion Syndrome 11 Cornelia de Lange Syndrome I (CDLS) 41 16p11.2-p12.2 microduplication Syndrome Potocki-Lupski Syndrome (17p11.2 duplication 12 Alpha Thalassemia, Mental Retardation Syndrome 42 Syndrome) 13 SIM1 43 Smith-Magenis Syndrome 14 Saethre-Chotzen Syndrome (SCS) 44 17q21.31 deletion Syndrome 15 8p23.1 duplication Syndrome 45 17q21.31 duplication Syndrome 16 8p23.1 deletion Syndrome 46 Holoprosencephaly type 4 (HPE4) 17 Trichorhinophalangeal type I Syndrome 47 Chromosome 18p deletion Syndrome 18 Branchlootorenal dysplasia Syndrome I 48 Duchenne/Becker mascular dystrophy (DMD/BMD) (BOR)/Melnick-Frazer Syndrome 19 Distal Arthrogryposis type 2B (DA2B) 49 Chromosome 18q deletion Syndrome 20 Langer-Giedion Syndrome (LGS) 50 Holoprosencephaly type 1 (HPE1) 21 Monosomy -

Chromosomal Microarray Chromosomal Microarray
Chromosomal Microarray Chromosomal Microarray Chromosomal microarray using array comparative genomic hybridization with single nucleotide polymorphism (array CGH+SNP) testing is a comprehensive analysis to examine the entire genome for deletions and duplications as well as copy neutral changes that may be clinically significant. This cutting-edge technology can be used in the aid of diagnosing prenatal and postnatal cases, including products of conception (POC). The array CGH technology uses dosage analysis of your patient’s DNA in comparison to a standardized reference DNA at loci all across the genome. Our comprehensive platform coupled with the expertise of our ABMGG-certified laboratory directors will give you the most accurate results possible. The microarray that we offer utilizes~180,000 CGH+SNP probes covering the entire genome with enriched coverage in regions known to be involved in submicroscopic chromosome abnormalities. This array yields high resolution detection of deletions and duplications with aberrations < 1kb possible in known genomic disease loci. The additional SNP probes allow for the simultaneous detection of regions of homozygosity for detection of uniparental disomy (UPD) and large regions with copy neutral absence of heterozygosity (AOH). What are the benefits of array CGH+SNP technology? • Diagnose children with developmental delay, intellectual • Higher resolution than conventional karyotyping to disability, and/or multiple congenital anomalie identify pathogenic deletions and duplications • Identify a specific -
FISH Mapping of the Sex-Reversal Region on Human Chromosome 9P in Two XY Females and in Primates
European Journal of Human Genetics (2000) 8, 167–173 © 2000 Macmillan Publishers Ltd All rights reserved 1018–4813/00 $15.00 y www.nature.com/ejhg ARTICLE FISH mapping of the sex-reversal region on human chromosome 9p in two XY females and in primates Zhihong Shan1,2, Bernhard Zabel3, Udo Trautmann4, Ulrike Hillig5, Chris Ottolenghi6, Yayu Wang1,2 and Thomas Haaf1 1Max-Planck-Institut f¨ur Molekulare Genetik, Berlin, Germany; 2Yunnan Center for Medical Genetics, People’s Hospital of Yunnan Province, Kunming, China; 3Kinderklinik, Johannes Gutenberg-Universit¨at Mainz; 4Institut f¨ur Humangenetik, Friedrich Alexander Universit¨at Erlangen-N¨urnberg; 5Zentrum f¨ur Humangenetik, Philipps-Universit¨at Marburg, Germany; 6Institut Pasteur, Paris, France Accumulating evidence suggests that haploinsufficiency of a dosage-sensitive gene(s) in human chromosome 9p24.3 is responsible for the failure of testicular development and feminisation in XY patients with monosomy for 9p. We have used molecular cytogenetic methods to characterise the sex-reversing 9p deletions in two XY females. Fluorescence in situ hybridisation (FISH) with YACs from the critical 9p region containing an evolutionarily conserved sex-determining gene, DMRT1, is a very fast and reliable assay for patient screening. Comparative YAC mapping on great ape and Old and New World monkey chromosomes demonstrated that the critical region was moved from an interstitial position on the ancestral primate chromosome to a very subtelomeric position in chimpanzee and humans by a pericentric inversion(s). Pathological 9p rearrangements may be the consequence of an evolutionary chromosome breakpoint in close proximity to the sex-reversal region. European Journal of Human Genetics (2000) 8, 167–173.