S41467-020-19704-X.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Temporal Proteomic Analysis of HIV Infection Reveals Remodelling of The
1 1 Temporal proteomic analysis of HIV infection reveals 2 remodelling of the host phosphoproteome 3 by lentiviral Vif variants 4 5 Edward JD Greenwood 1,2,*, Nicholas J Matheson1,2,*, Kim Wals1, Dick JH van den Boomen1, 6 Robin Antrobus1, James C Williamson1, Paul J Lehner1,* 7 1. Cambridge Institute for Medical Research, Department of Medicine, University of 8 Cambridge, Cambridge, CB2 0XY, UK. 9 2. These authors contributed equally to this work. 10 *Correspondence: [email protected]; [email protected]; [email protected] 11 12 Abstract 13 Viruses manipulate host factors to enhance their replication and evade cellular restriction. 14 We used multiplex tandem mass tag (TMT)-based whole cell proteomics to perform a 15 comprehensive time course analysis of >6,500 viral and cellular proteins during HIV 16 infection. To enable specific functional predictions, we categorized cellular proteins regulated 17 by HIV according to their patterns of temporal expression. We focussed on proteins depleted 18 with similar kinetics to APOBEC3C, and found the viral accessory protein Vif to be 19 necessary and sufficient for CUL5-dependent proteasomal degradation of all members of the 20 B56 family of regulatory subunits of the key cellular phosphatase PP2A (PPP2R5A-E). 21 Quantitative phosphoproteomic analysis of HIV-infected cells confirmed Vif-dependent 22 hyperphosphorylation of >200 cellular proteins, particularly substrates of the aurora kinases. 23 The ability of Vif to target PPP2R5 subunits is found in primate and non-primate lentiviral 2 24 lineages, and remodeling of the cellular phosphoproteome is therefore a second ancient and 25 conserved Vif function. -

Novelty Indicator for Enhanced Prioritization of Predicted Gene Ontology Annotations
IEEE/ACM TRANSACTIONS ON COMPUTATIONAL BIOLOGY AND BIOINFORMATICS, VOL. X, NO. X, MONTHXXX 20XX 1 Novelty Indicator for Enhanced Prioritization of Predicted Gene Ontology Annotations Davide Chicco, Fernando Palluzzi, and Marco Masseroli Abstract—Biomolecular controlled annotations have become pivotal in computational biology, because they allow scientists to analyze large amounts of biological data to better understand their test results, and to infer new knowledge. Yet, biomolecular annotation databases are incomplete by definition, like our knowledge of biology, and may contain errors and inconsistent information. In this context, machine-learning algorithms able to predict and prioritize new biomolecular annotations are both effective and efficient, especially if compared with the time-consuming trials of biological validation. To limit the possibility that these techniques predict obvious and trivial high-level features, and to help prioritizing their results, we introduce here a new element that can improve the accuracy and relevance of the results of an annotation prediction and prioritization pipeline. We propose a novelty indicator able to state the level of ”newness” (or ”originality”) of the annotations predicted for a specific gene to Gene Ontology terms, and to help prioritizing the most novel and interesting annotations predicted. We performed a thorough biological functional analysis of the prioritized annotations predicted with high accuracy by using this indicator and our previously proposed prediction algorithms. The relevance -

Loss of PPP2R2A Inhibits Homologous Recombination DNA Repair and Predicts
Author Manuscript Published OnlineFirst on October 18, 2012; DOI: 10.1158/0008-5472.CAN-12-1667 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Loss of PPP2R2A inhibits homologous recombination DNA repair and predicts tumor sensitivity to PARP inhibition Peter Kalev1, Michal Simicek1, Iria Vazquez1, Sebastian Munck1, Liping Chen2, Thomas Soin1, Natasha Danda1, Wen Chen2 and Anna Sablina1,* 1VIB Center for the Biology of Disease; Center for Human Genetics, KULeuven, Leuven 3000 Belgium 2Department of Toxicology, Faculty of Preventive Medicine, Guangdong Provincial Key Laboratory of Food, Nutrition and Health, School of Public Health, Sun Yat-sen University, Guangzhou 510080, China *Corresponding author information: [email protected] Contact information: Anna A. Sablina, Ph.D. CME Department, KULeuven O&N I Herestraat 49, bus 602 Leuven, Belgium 3000 Tel: +3216330790 Fax: +3216330145 Running title: The role of PPP2R2A in DNA repair Keywords: PP2A, ATM, DNA repair, cancer, PARP inhibition Conflict of interests: The authors claim no conflict of interest. - 1 - Downloaded from cancerres.aacrjournals.org on September 27, 2021. © 2012 American Association for Cancer Research. Author Manuscript Published OnlineFirst on October 18, 2012; DOI: 10.1158/0008-5472.CAN-12-1667 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Abstract Reversible phosphorylation plays a critical role in DNA repair. Here we report the results of a loss-of-function screen that identifies the PP2A heterotrimeric serine/threonine phosphatases PPP2R2A, PPP2R2D, PPP2R5A and PPP2R3C in double-strand break (DSB) repair. In particular, we found that PPP2R2A-containing complexes directly dephosphorylated ATM at S367, S1893, and S1981 to regulate its retention at DSB sites. -

A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family
Rockefeller University Digital Commons @ RU Student Theses and Dissertations 2018 A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family Linda Molla Follow this and additional works at: https://digitalcommons.rockefeller.edu/ student_theses_and_dissertations Part of the Life Sciences Commons A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY A Thesis Presented to the Faculty of The Rockefeller University in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy by Linda Molla June 2018 © Copyright by Linda Molla 2018 A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY Linda Molla, Ph.D. The Rockefeller University 2018 APOBEC2 is a member of the AID/APOBEC cytidine deaminase family of proteins. Unlike most of AID/APOBEC, however, APOBEC2’s function remains elusive. Previous research has implicated APOBEC2 in diverse organisms and cellular processes such as muscle biology (in Mus musculus), regeneration (in Danio rerio), and development (in Xenopus laevis). APOBEC2 has also been implicated in cancer. However the enzymatic activity, substrate or physiological target(s) of APOBEC2 are unknown. For this thesis, I have combined Next Generation Sequencing (NGS) techniques with state-of-the-art molecular biology to determine the physiological targets of APOBEC2. Using a cell culture muscle differentiation system, and RNA sequencing (RNA-Seq) by polyA capture, I demonstrated that unlike the AID/APOBEC family member APOBEC1, APOBEC2 is not an RNA editor. Using the same system combined with enhanced Reduced Representation Bisulfite Sequencing (eRRBS) analyses I showed that, unlike the AID/APOBEC family member AID, APOBEC2 does not act as a 5-methyl-C deaminase. -

Therapeutic Re-Activation of Protein Phosphatase 2A in Acute Myeloid
MINI REVIEW ARTICLE published: 02 February 2015 doi: 10.3389/fonc.2015.00016 Therapeutic re-activation of protein phosphatase 2A in acute myeloid leukemia Kavitha Ramaswamy, Barbara Spitzer and Alex Kentsis* Molecular Pharmacology and Chemistry Program, Department of Pediatrics, Sloan Kettering Institute, Memorial Sloan Kettering Cancer Center, Weill Medical College of Cornell University, New York, NY, USA Edited by: Protein phosphatase 2A (PP2A) is a serine/threonine phosphatase that is required for nor- Peter Ruvolo, The University of Texas mal cell growth and development. PP2A is a potent tumor suppressor, which is inactivated MD Anderson Cancer Center, USA in cancer cells as a result of genetic deletions and mutations. In myeloid leukemias, genes Reviewed by: encoding PP2A subunits are generally intact. Instead, PP2A is functionally inhibited by Peter Ruvolo, The University of Texas MD Anderson Cancer Center, USA post-translational modifications of its catalytic C subunit, and interactions with negative Alejandro Gutierrez, Boston Children’s regulators by its regulatory B and scaffold A subunits. Here, we review the molecular Hospital, USA mechanisms of genetic and functional inactivation of PP2A in human cancers, with a par- *Correspondence: ticular focus on human acute myeloid leukemias (AML). By analyzing expression of genes Alex Kentsis, Molecular Pharmacology and Chemistry encoding PP2A subunits using transcriptome sequencing, we find that PP2A dysregulation Program, Department of Pediatrics, in AML is characterized by silencing -

Live-Cell Imaging Rnai Screen Identifies PP2A–B55α and Importin-Β1 As Key Mitotic Exit Regulators in Human Cells
LETTERS Live-cell imaging RNAi screen identifies PP2A–B55α and importin-β1 as key mitotic exit regulators in human cells Michael H. A. Schmitz1,2,3, Michael Held1,2, Veerle Janssens4, James R. A. Hutchins5, Otto Hudecz6, Elitsa Ivanova4, Jozef Goris4, Laura Trinkle-Mulcahy7, Angus I. Lamond8, Ina Poser9, Anthony A. Hyman9, Karl Mechtler5,6, Jan-Michael Peters5 and Daniel W. Gerlich1,2,10 When vertebrate cells exit mitosis various cellular structures can contribute to Cdk1 substrate dephosphorylation during vertebrate are re-organized to build functional interphase cells1. This mitotic exit, whereas Ca2+-triggered mitotic exit in cytostatic-factor- depends on Cdk1 (cyclin dependent kinase 1) inactivation arrested egg extracts depends on calcineurin12,13. Early genetic studies in and subsequent dephosphorylation of its substrates2–4. Drosophila melanogaster 14,15 and Aspergillus nidulans16 reported defects Members of the protein phosphatase 1 and 2A (PP1 and in late mitosis of PP1 and PP2A mutants. However, the assays used in PP2A) families can dephosphorylate Cdk1 substrates in these studies were not specific for mitotic exit because they scored pro- biochemical extracts during mitotic exit5,6, but how this relates metaphase arrest or anaphase chromosome bridges, which can result to postmitotic reassembly of interphase structures in intact from defects in early mitosis. cells is not known. Here, we use a live-cell imaging assay and Intracellular targeting of Ser/Thr phosphatase complexes to specific RNAi knockdown to screen a genome-wide library of protein substrates is mediated by a diverse range of regulatory and targeting phosphatases for mitotic exit functions in human cells. We subunits that associate with a small group of catalytic subunits3,4,17. -

Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors
University of Cincinnati Date: 12/20/2010 I, Arturo R Maldonado , hereby submit this original work as part of the requirements for the degree of Doctor of Philosophy in Developmental Biology. It is entitled: Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors Student's name: Arturo R Maldonado This work and its defense approved by: Committee chair: Jeffrey Whitsett Committee member: Timothy Crombleholme, MD Committee member: Dan Wiginton, PhD Committee member: Rhonda Cardin, PhD Committee member: Tim Cripe 1297 Last Printed:1/11/2011 Document Of Defense Form Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors A dissertation submitted to the Graduate School of the University of Cincinnati College of Medicine in partial fulfillment of the requirements for the degree of DOCTORATE OF PHILOSOPHY (PH.D.) in the Division of Molecular & Developmental Biology 2010 By Arturo Rafael Maldonado B.A., University of Miami, Coral Gables, Florida June 1993 M.D., New Jersey Medical School, Newark, New Jersey June 1999 Committee Chair: Jeffrey A. Whitsett, M.D. Advisor: Timothy M. Crombleholme, M.D. Timothy P. Cripe, M.D. Ph.D. Dan Wiginton, Ph.D. Rhonda D. Cardin, Ph.D. ABSTRACT Since 1999, cancer has surpassed heart disease as the number one cause of death in the US for people under the age of 85. Malignant Peripheral Nerve Sheath Tumor (MPNST), a common malignancy in patients with Neurofibromatosis, and colorectal cancer are midkine- producing tumors with high mortality rates. In vitro and preclinical xenograft models of MPNST were utilized in this dissertation to study the role of midkine (MDK), a tumor-specific gene over- expressed in these tumors and to test the efficacy of a MDK-transcriptionally targeted oncolytic HSV-1 (oHSV). -

Loss of the E3 Ubiquitin Ligase MKRN1 Represses Diet-Induced Metabolic Syndrome Through AMPK Activation
ARTICLE DOI: 10.1038/s41467-018-05721-4 OPEN Loss of the E3 ubiquitin ligase MKRN1 represses diet-induced metabolic syndrome through AMPK activation Min-Sik Lee1, Hyun-Ji Han2, Su Yeon Han2, Il Young Kim3,4, Sehyun Chae5, Choong-Sil Lee2, Sung Eun Kim2, Seul Gi Yoon4, Jun-Won Park4, Jung-Hoon Kim2, Soyeon Shin2, Manhyung Jeong2, Aram Ko2, Ho-Young Lee6, Kyoung-Jin Oh 7, Yun-Hee Lee 8, Kwang-Hee Bae7, Seung-Hoi Koo9, Jea-woo Kim10, Je Kyung Seong3,4, Daehee Hwang5 & Jaewhan Song 2 1234567890():,; AMP-activated protein kinase (AMPK) plays a key role in controlling energy metabolism in response to physiological and nutritional status. Although AMPK activation has been pro- posed as a promising molecular target for treating obesity and its related comorbidities, the use of pharmacological AMPK activators has been met with contradictory therapeutic challenges. Here we show a regulatory mechanism for AMPK through its ubiquitination and degradation by the E3 ubiquitin ligase makorin ring finger protein 1 (MKRN1). MKRN1 depletion promotes glucose consumption and suppresses lipid accumulation due to AMPK stabilisation and activation. Accordingly, MKRN1-null mice show chronic AMPK activation in both liver and adipose tissue, resulting in significant suppression of diet-induced metabolic syndrome. We demonstrate also its therapeutic effect by administering shRNA targeting MKRN1 into obese mice that reverses non-alcoholic fatty liver disease. We suggest that ubiquitin-dependent AMPK degradation represents a target therapeutic strategy for meta- bolic disorders. 1 Harvard Medical School, Boston Children’s Hospital, 3 Blackfan Circle CLS-16060.2, Boston, MA 02115, USA. 2 Department of Biochemistry, College of Life Science and Biotechnology, Yonsei University, Seoul 03722, Republic of Korea. -

Prioritizing and Characterizing Functionally Relevant Genes Across Human Tissues
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.22.427774; this version posted January 22, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Prioritizing and characterizing functionally relevant genes across human tissues Authors Gowthami Somepalli1†, Sarthak Sahoo2†, Arashdeep Singh3, Sridhar Hannenhalli3* Affiliations 1 Center for Bioinformatics and Computational Biology, University of Maryland, College Park, MD, USA 2 Undergraduate program, Indian Institute of Science, Bengaluru, India 3 Cancer Data Science Lab, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA † These authors contributed equally to this work *Corresponding author. Email: [email protected] (S.H.) Abstract Knowledge of genes that are critical to a tissue’s function remains difficult to ascertain and presents a major bottleneck toward a mechanistic understanding of genotype-phenotype links. Here, we present the first machine learning model – FUGUE – combining transcriptional and network features, to predict tissue-relevant genes across 30 human tissues. FUGUE achieves an average cross-validation auROC of 0.86 and auPRC of 0.50 (expected 0.09). In three independent datasets, FUGUE accurately distinguishes tissue or cell type-specific genes, significantly outperforming the conventional metric based on tissue-specific expression alone. Comparison of tissue-relevant transcription factors across tissue recapitulate their developmental relationships. Interestingly, the tissue-relevant genes cluster on the genome within topologically associated domains and furthermore, are highly enriched for differentially expressed genes in the corresponding cancer type. -

Identification of a 6-Cm Minimal Deletion at 11Q23.1–23.2 and Exclusion of PPP2R1B Gene As a Deletion Target in Cervical Cancer1
[CANCER RESEARCH 60, 6677–6682, December 1, 2000] Identification of a 6-cM Minimal Deletion at 11q23.1–23.2 and Exclusion of PPP2R1B Gene as a Deletion Target in Cervical Cancer1 Hugo Arias Pulido, Mohamed J. Fakruddin, Anupam Chatterjee, Edward D. Esplin, Nestor Belen˜o, Gilberto Martı´nez, Hector Posso, Glen A. Evans, and V. V. V. S. Murty2 Department of Pathology, College of Physicians and Surgeons of Columbia University, New York, New York 10032 [H. A. P., M. J. F., A. C., V. V. V. S. M.]; Departments of Genetics, Pathology, Gynecology, and Epidemiology, Instituto Nacional de Cancerologı´a, Santa Fe de Bogota´, Colombia [H. A. P., N. B., G. M., H. P.]; Southwestern Medical Center, Dallas, Texas 75390 [E. D. E., G. A. E.]; and Department of Zoology, North-Eastern Hill University, Shillong, 793022 India [A. C.] ABSTRACT data suggest that only a certain fraction of HPV-infected CIN lesions progress to invasive CCs with variable latency periods (2–4). These Previous functional and deletion mapping studies on cervical cancer data, therefore, suggest that additional genetic alterations may be (CC) have implicated one or more tumor suppressor genes (TSGs) on necessary for the progression of CCs. Delineation of such genetic chromosome 11 at q13 and q22–24 regions. Of these, the 11q22–24 region exhibits frequent allelic deletions in a variety of solid tumor types, sug- changes may be of relevance in understanding cervical carcinogenesis gesting the presence of critical genes for tumor suppression in this region. and will have implications in early detection and identification of However, the precise region of deletion on 11q is not clearly defined in CC. -
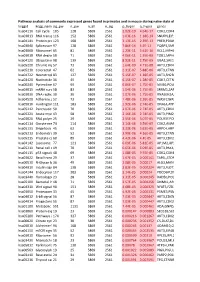
Pathway Analysis of Commonly Expressed Genes Found in Primates and in Mouse During Naïve State of Pluripotenc Keggid Kegg Names
Pathway analysis of commonly expressed genes found in primates and in mouse during naïve state of pluripotency. keggid kegg_namesig_pw n_pw n_all n_sig p_hyper q_hyper genes hsa04110 Cell cycle 105 128 5869 2561 2.02E-19 4.64E-17 CDK2,CDK4,CDK6,CDK7,CDKN1A,CDKN1B,STAG1,CDKN2B,ANAPC10,MAD2L2,STAG2,GADD45G,DBF4,YWHAQ,CHEK1,CHEK2,CREBBP,GADD45A,E2F1,E2F3,E2F4,E2F5,EP300,ORC3,CDC26,ABL1,ANAPC13,SFN,GSK3B,ANAPC2,ANAPC4,HDAC1,HDAC2,MAD2L1,SMAD2,SMAD3,SMAD4,MCM2,MCM3,MCM4,MCM5,MCM6,MCM7,MDM2,MYC,GADD45B,ATM,ORC1,ORC2,ORC4,ORC5,FZR1,ANAPC7,ANAPC11,PLK1,ATR,PRKDC,RAD21,RB1,RBL1,CCND1,ANAPC1,SKP2,BUB1,BUB1B,TFDP1,TFDP2,TGFB1,TGFB2,TTK,WEE1,YWHAB,YWHAE,YWHAG,YWHAH,YWHAZ,ZBTB17,SMC1A,CDC7,CDC45,MAD1L1,CDC14A,CDC23,CDC16,CCNA2,CCNB1,CCND2,CCND3,CCNE1,CCNH,PKMYT1,SMC3,CCNB2,CCNE2,BUB3,PTTG1,ESPL1,CDK1,CDC6,CDC20,CDC25A,CDC25B,CDC25C,CDC27,RBX1 hsa03013 RNA transport116 152 5869 2561 1.03E-16 1.18E-14 SNUPN,EIF1,POP7,SRRM1,SAP18,EIF1B,PRMT5,TACC3,NXF1,RPP30,RPP38,PAIP1,POP4,RPP40,RNPS1,POP1,STRAP,DDX20,XPOT,CLNS1A,NUP35,RPP25L,EEF1A1,EEF1A2,EIF2S1,EIF2B1,EIF4A1,EIF4B,EIF4E,EIF4EBP1,EIF4G1,EIF4G2,EIF5,CASC3,NCBP2,ACIN1,NUP205,NUP210,NUP62,GEMIN5,UPF2,PABPC1,NXT1,NUP43,EIF3E,KPNB1,MAGOH,NCBP1,NUP88,NUP98,GEMIN4,NMD3,POP5,PHAX,NUP54,PNN,RPP25,GEMIN8,MAGOHB,ELAC1,NDC1,NUP133,NXT2,NUP107,THOC2,XPO5,RAN,RANBP2,RANGAP1,SENP2,UPF1,ELAC2,SEC13,UPF3B,SMN1,SUMO3,SUMO2,TPR,SUMO1,XPO1,NUP37,DDX39B,THOC6,GEMIN7,GEMIN6,RPP21,NUP85,THOC7,NUP214,AAAS,SEH1L,THOC3,RAE1,THOC5,EIF3A,EIF3B,EIF3C,EIF3D,EIF3F,EIF3G,EIF3H,EIF3I,EIF4G3,PABPC4,EIF2B4,EIF2B2,EIF2B5,EIF2S2,EIF4E2,NUP155,EIF5B,TGS1,NUP93,EIF4A3,NUP153,THOC1 -

Protein Phosphatase 2A Regulatory Subunits and Cancer
Biochimica et Biophysica Acta 1795 (2009) 1–15 Contents lists available at ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbacan Review Protein phosphatase 2A regulatory subunits and cancer Pieter J.A. Eichhorn 1, Menno P. Creyghton 2, René Bernards ⁎ Division of Molecular Carcinogenesis, Center for Cancer Genomics and Center for Biomedical Genetics, The Netherlands Cancer Institute, Plesmanlaan 121, 1066 CX Amsterdam, The Netherlands article info abstract Article history: The serine/threonine protein phosphatase (PP2A) is a trimeric holoenzyme that plays an integral role in the Received 7 April 2008 regulation of a number of major signaling pathways whose deregulation can contribute to cancer. The Received in revised form 20 May 2008 specificity and activity of PP2A are highly regulated through the interaction of a family of regulatory B Accepted 21 May 2008 subunits with the substrates. Accumulating evidence indicates that PP2A acts as a tumor suppressor. In this Available online 3 June 2008 review we summarize the known effects of specific PP2A holoenzymes and their roles in cancer relevant pathways. In particular we highlight PP2A function in the regulation of MAPK and Wnt signaling. Keywords: Protein phosphatase 2A © 2008 Elsevier B.V. All rights reserved. Signal transduction Cancer Contents 1. Introduction ............................................................... 1 2. PP2A structure and function ....................................................... 2 2.1. The catalytic subunit (PP2Ac).................................................... 2 2.2. The structural subunit (PR65) ................................................... 3 2.3. The regulatory B subunits ..................................................... 3 2.3.1. The B/PR55 family of B subunits .............................................. 3 2.3.2. The B′/PR61 family of β subunits ............................................. 4 2.3.3. The B″/PR72 family of β subunits ............................................