A Cross-Platform Analysis of 14,177 Expression Quantitative Trait Loci Derived from Lymphoblastoid Cell Lines
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
![RT² Profiler PCR Array (96-Well Format and 384-Well [4 X 96] Format)](https://docslib.b-cdn.net/cover/1030/rt%C2%B2-profiler-pcr-array-96-well-format-and-384-well-4-x-96-format-301030.webp)
RT² Profiler PCR Array (96-Well Format and 384-Well [4 X 96] Format)
RT² Profiler PCR Array (96-Well Format and 384-Well [4 x 96] Format) Human Mitochondria Cat. no. 330231 PAHS-087ZA For pathway expression analysis Format For use with the following real-time cyclers RT² Profiler PCR Array, Applied Biosystems® models 5700, 7000, 7300, 7500, Format A 7700, 7900HT, ViiA™ 7 (96-well block); Bio-Rad® models iCycler®, iQ™5, MyiQ™, MyiQ2; Bio-Rad/MJ Research Chromo4™; Eppendorf® Mastercycler® ep realplex models 2, 2s, 4, 4s; Stratagene® models Mx3005P®, Mx3000P®; Takara TP-800 RT² Profiler PCR Array, Applied Biosystems models 7500 (Fast block), 7900HT (Fast Format C block), StepOnePlus™, ViiA 7 (Fast block) RT² Profiler PCR Array, Bio-Rad CFX96™; Bio-Rad/MJ Research models DNA Format D Engine Opticon®, DNA Engine Opticon 2; Stratagene Mx4000® RT² Profiler PCR Array, Applied Biosystems models 7900HT (384-well block), ViiA 7 Format E (384-well block); Bio-Rad CFX384™ RT² Profiler PCR Array, Roche® LightCycler® 480 (96-well block) Format F RT² Profiler PCR Array, Roche LightCycler 480 (384-well block) Format G RT² Profiler PCR Array, Fluidigm® BioMark™ Format H Sample & Assay Technologies Description The Human Mitochondria RT² Profiler PCR Array profiles the expression of 84 genes involved in the biogenesis and function of the cell's powerhouse organelle. The genes monitored by this array include regulators and mediators of mitochondrial molecular transport of not only the metabolites needed for the electron transport chain and oxidative phosphorylation, but also the ions required for maintaining the mitochondrial membrane polarization and potential important for ATP synthesis. Metabolism and energy production are not the only functions of mitochondria. -

Human CLPB) Is a Potent Mitochondrial Protein Disaggregase That Is Inactivated By
bioRxiv preprint doi: https://doi.org/10.1101/2020.01.17.911016; this version posted January 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Skd3 (human CLPB) is a potent mitochondrial protein disaggregase that is inactivated by 3-methylglutaconic aciduria-linked mutations Ryan R. Cupo1,2 and James Shorter1,2* 1Department of Biochemistry and Biophysics, 2Pharmacology Graduate Group, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA 19104, U.S.A. *Correspondence: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.01.17.911016; this version posted January 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. ABSTRACT Cells have evolved specialized protein disaggregases to reverse toxic protein aggregation and restore protein functionality. In nonmetazoan eukaryotes, the AAA+ disaggregase Hsp78 resolubilizes and reactivates proteins in mitochondria. Curiously, metazoa lack Hsp78. Hence, whether metazoan mitochondria reactivate aggregated proteins is unknown. Here, we establish that a mitochondrial AAA+ protein, Skd3 (human CLPB), couples ATP hydrolysis to protein disaggregation and reactivation. The Skd3 ankyrin-repeat domain combines with conserved AAA+ elements to enable stand-alone disaggregase activity. A mitochondrial inner-membrane protease, PARL, removes an autoinhibitory peptide from Skd3 to greatly enhance disaggregase activity. Indeed, PARL-activated Skd3 dissolves α-synuclein fibrils connected to Parkinson’s disease. Human cells lacking Skd3 exhibit reduced solubility of various mitochondrial proteins, including anti-apoptotic Hax1. -

Skd3 (Human CLPB) Is a Potent Mitochondrial Protein Disaggregase That Is Inactivated By
bioRxiv preprint first posted online Jan. 18, 2020; doi: http://dx.doi.org/10.1101/2020.01.17.911016. The copyright holder for this preprint (which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Skd3 (human CLPB) is a potent mitochondrial protein disaggregase that is inactivated by 3-methylglutaconic aciduria-linked mutations Ryan R. Cupo1,2 and James Shorter1,2* 1Department of Biochemistry and Biophysics, 2Pharmacology Graduate Group, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA 19104, U.S.A. *Correspondence: [email protected] 1 bioRxiv preprint first posted online Jan. 18, 2020; doi: http://dx.doi.org/10.1101/2020.01.17.911016. The copyright holder for this preprint (which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. ABSTRACT Cells have evolved specialized protein disaggregases to reverse toxic protein aggregation and restore protein functionality. In nonmetazoan eukaryotes, the AAA+ disaggregase Hsp78 resolubilizes and reactivates proteins in mitochondria. Curiously, metazoa lack Hsp78. Hence, whether metazoan mitochondria reactivate aggregated proteins is unknown. Here, we establish that a mitochondrial AAA+ protein, Skd3 (human CLPB), couples ATP hydrolysis to protein disaggregation and reactivation. The Skd3 ankyrin-repeat domain combines with conserved AAA+ elements to enable stand-alone disaggregase activity. A mitochondrial inner-membrane protease, PARL, removes an autoinhibitory peptide from Skd3 to greatly enhance disaggregase activity. Indeed, PARL-activated Skd3 dissolves α-synuclein fibrils connected to Parkinson’s disease. -
![RT² Profiler PCR Array (96-Well Format and 384-Well [4 X 96] Format)](https://docslib.b-cdn.net/cover/1423/rt%C2%B2-profiler-pcr-array-96-well-format-and-384-well-4-x-96-format-2611423.webp)
RT² Profiler PCR Array (96-Well Format and 384-Well [4 X 96] Format)
RT² Profiler PCR Array (96-Well Format and 384-Well [4 x 96] Format) Rat Mitochondria Cat. no. 330231 PARN-087ZA For pathway expression analysis Format For use with the following real-time cyclers RT² Profiler PCR Array, Applied Biosystems® models 5700, 7000, 7300, 7500, Format A 7700, 7900HT, ViiA™ 7 (96-well block); Bio-Rad® models iCycler®, iQ™5, MyiQ™, MyiQ2; Bio-Rad/MJ Research Chromo4™; Eppendorf® Mastercycler® ep realplex models 2, 2s, 4, 4s; Stratagene® models Mx3005P®, Mx3000P®; Takara TP-800 RT² Profiler PCR Array, Applied Biosystems models 7500 (Fast block), 7900HT (Fast Format C block), StepOnePlus™, ViiA 7 (Fast block) RT² Profiler PCR Array, Bio-Rad CFX96™; Bio-Rad/MJ Research models DNA Format D Engine Opticon®, DNA Engine Opticon 2; Stratagene Mx4000® RT² Profiler PCR Array, Applied Biosystems models 7900HT (384-well block), ViiA 7 Format E (384-well block); Bio-Rad CFX384™ RT² Profiler PCR Array, Roche® LightCycler® 480 (96-well block) Format F RT² Profiler PCR Array, Roche LightCycler 480 (384-well block) Format G RT² Profiler PCR Array, Fluidigm® BioMark™ Format H Sample & Assay Technologies Description The Rat Mitochondria RT² Profiler PCR Array profiles the expression of 84 genes involved in the biogenesis and function of the cell's powerhouse organelle. The genes monitored by this array include regulators and mediators of mitochondrial molecular transport of not only the metabolites needed for the electron transport chain and oxidative phosphorylation, but also the ions required for maintaining the mitochondrial membrane polarization and potential important for ATP synthesis. Metabolism and energy production are not the only functions of mitochondria. -

Spatial Sorting Enables Comprehensive Characterization of Liver Zonation
ARTICLES https://doi.org/10.1038/s42255-019-0109-9 Spatial sorting enables comprehensive characterization of liver zonation Shani Ben-Moshe1,3, Yonatan Shapira1,3, Andreas E. Moor 1,2, Rita Manco1, Tamar Veg1, Keren Bahar Halpern1 and Shalev Itzkovitz 1* The mammalian liver is composed of repeating hexagonal units termed lobules. Spatially resolved single-cell transcriptomics has revealed that about half of hepatocyte genes are differentially expressed across the lobule, yet technical limitations have impeded reconstructing similar global spatial maps of other hepatocyte features. Here, we show how zonated surface markers can be used to sort hepatocytes from defined lobule zones with high spatial resolution. We apply transcriptomics, microRNA (miRNA) array measurements and mass spectrometry proteomics to reconstruct spatial atlases of multiple zon- ated features. We demonstrate that protein zonation largely overlaps with messenger RNA zonation, with the periportal HNF4α as an exception. We identify zonation of miRNAs, such as miR-122, and inverse zonation of miRNAs and their hepa- tocyte target genes, highlighting potential regulation of gene expression levels through zonated mRNA degradation. Among the targets, we find the pericentral Wingless-related integration site (Wnt) receptors Fzd7 and Fzd8 and the periportal Wnt inhibitors Tcf7l1 and Ctnnbip1. Our approach facilitates reconstructing spatial atlases of multiple cellular features in the liver and other structured tissues. he mammalian liver is a structured organ, consisting of measurements would broaden our understanding of the regulation repeating hexagonally shaped units termed ‘lobules’ (Fig. 1a). of liver zonation and could be used to model liver metabolic func- In mice, each lobule consists of around 9–12 concentric lay- tion more precisely. -

Foot Malformation and Long-Bone Deficiency (SHFLD)
European Journal of Human Genetics (2011) 19, 1144–1151 & 2011 Macmillan Publishers Limited All rights reserved 1018-4813/11 www.nature.com/ejhg ARTICLE 17p13.3 microduplications are associated with split-hand/foot malformation and long-bone deficiency (SHFLD) Christine M Armour*,1, Dennis E Bulman2,3, Olga Jarinova4, Richard Curtis Rogers5, Kate B Clarkson5, Barbara R DuPont5, Alka Dwivedi5, Frank O Bartel5, Laura McDonell2,6, Charles E Schwartz5,7, Kym M Boycott2,8, David B Everman*,5,9 and Gail E Graham2,8,9 Split-hand/foot malformation with long-bone deficiency (SHFLD) is a relatively rare autosomal-dominant skeletal disorder, characterized by variable expressivity and incomplete penetrance. Although several chromosomal loci for SHFLD have been identified, the molecular basis and pathogenesis of most SHFLD cases are unknown. In this study we describe three unrelated kindreds, in which SHFLD segregated with distinct but overlapping duplications in 17p13.3, a region previously linked to SHFLD. In a large three-generation family, the disorder was found to segregate with a 254 kb microduplication; a second microduplication of 527 kb was identified in an affected female and her unaffected mother, and a 430 kb microduplication versus microtriplication was identified in three affected members of a multi-generational family. These findings, along with previously published data, suggest that one locus responsible for this form of SHFLD is located within a 173 kb overlapping critical region, and that the copy gains are incompletely penetrant. European Journal of Human Genetics (2011) 19, 1144–1151; doi:10.1038/ejhg.2011.97; published online 1 June 2011 Keywords: split-hand/foot malformation; SHFM; SHFLD; microduplication; microarray; conserved regulatory element INTRODUCTION dystrophy (EEM) syndrome5 (OMIM # 225 280). -

Mitochondrial Protein Translocases for Survival and Wellbeing
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector FEBS Letters 588 (2014) 2484–2495 journal homepage: www.FEBSLetters.org Review Mitochondrial protein translocases for survival and wellbeing Anna Magdalena Sokol a, Malgorzata Eliza Sztolsztener a, Michal Wasilewski a, Eva Heinz b,c, ⇑ Agnieszka Chacinska a, a Laboratory of Mitochondrial Biogenesis, International Institute of Molecular and Cell Biology, Warsaw 02-109, Poland b Department of Microbiology, Monash University, Melbourne, Victoria 3800, Australia c Victorian Bioinformatics Consortium, Monash University, Melbourne, Victoria 3800, Australia article info abstract Article history: Mitochondria are involved in many essential cellular activities. These broad functions explicate the Received 28 April 2014 need for the well-orchestrated biogenesis of mitochondrial proteins to avoid death and pathological Revised 15 May 2014 consequences, both in unicellular and more complex organisms. Yeast as a model organism has Accepted 15 May 2014 been pivotal in identifying components and mechanisms that drive the transport and sorting of Available online 24 May 2014 nuclear-encoded mitochondrial proteins. The machinery components that are involved in the Edited by Wilhelm Just import of mitochondrial proteins are generally evolutionarily conserved within the eukaryotic king- dom. However, topological and functional differences have been observed. We review the similari- ties and differences in mitochondrial translocases from yeast to human. Additionally, we provide a Keywords: Cancer systematic overview of the contribution of mitochondrial import machineries to human patholo- HIF1a gies, including cancer, mitochondrial diseases, and neurodegeneration. Mitochondrial disease Ó 2014 Federation of European Biochemical Societies. Published by Elsevier B.V. -

146823088.Pdf
Syddansk Universitet Identification of genetic elements in metabolism by high-throughput mouse phenotyping ARTICLE DOI: 10.1038/s41467-017-01995-2 OPEN Identification of genetic elements in metabolism by high-throughput mouse phenotyping Jan Rozman et al.# Metabolic diseases are a worldwide problem but the underlying genetic factors and their relevance to metabolic disease remain incompletely understood. Genome-wide research is needed to characterize so-far unannotated mammalian metabolic genes. Here, we generate 1234567890 and analyze metabolic phenotypic data of 2016 knockout mouse strains under the aegis of the International Mouse Phenotyping Consortium (IMPC) and find 974 gene knockouts with strong metabolic phenotypes. 429 of those had no previous link to metabolism and 51 genes remain functionally completely unannotated. We compared human orthologues of these uncharacterized genes in five GWAS consortia and indeed 23 candidate genes are associated with metabolic disease. We further identify common regulatory elements in promoters of candidate genes. As each regulatory element is composed of several transcription factor binding sites, our data reveal an extensive metabolic phenotype-associated network of co- regulated genes. Our systematic mouse phenotype analysis thus paves the way for full functional annotation of the genome. Correspondence and requests for materials should be addressed to M.H.d.A. (email: [email protected]) #A full list of authors and their affliations appears at the end of the paper NATURE COMMUNICATIONS | (2018) 9:288 | DOI: 10.1038/s41467-017-01995-2 | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | DOI: 10.1038/s41467-017-01995-2 etabolic disorders, including obesity and type 2 diabetes results obtained from genotype–phenotype associations in disease Mmellitus, are major challenges for public health. -

Identification of Genetic Elements in Metabolism by High-Throughput
ARTICLE DOI: 10.1038/s41467-017-01995-2 OPEN Identification of genetic elements in metabolism by high-throughput mouse phenotyping Jan Rozman et al.# Metabolic diseases are a worldwide problem but the underlying genetic factors and their relevance to metabolic disease remain incompletely understood. Genome-wide research is needed to characterize so-far unannotated mammalian metabolic genes. Here, we generate 1234567890 and analyze metabolic phenotypic data of 2016 knockout mouse strains under the aegis of the International Mouse Phenotyping Consortium (IMPC) and find 974 gene knockouts with strong metabolic phenotypes. 429 of those had no previous link to metabolism and 51 genes remain functionally completely unannotated. We compared human orthologues of these uncharacterized genes in five GWAS consortia and indeed 23 candidate genes are associated with metabolic disease. We further identify common regulatory elements in promoters of candidate genes. As each regulatory element is composed of several transcription factor binding sites, our data reveal an extensive metabolic phenotype-associated network of co- regulated genes. Our systematic mouse phenotype analysis thus paves the way for full functional annotation of the genome. Correspondence and requests for materials should be addressed to M.H.d.A. (email: [email protected]) #A full list of authors and their affliations appears at the end of the paper NATURE COMMUNICATIONS | (2018) 9:288 | DOI: 10.1038/s41467-017-01995-2 | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | DOI: 10.1038/s41467-017-01995-2 etabolic disorders, including obesity and type 2 diabetes results obtained from genotype–phenotype associations in disease Mmellitus, are major challenges for public health. -

Chromosomal Instability and Copy Number Alterations in Barrett's Esophagus and Esophageal Adenocarcinoma Thomas G
Human Cancer Biology Chromosomal Instability and Copy Number Alterations in Barrett's Esophagus and Esophageal Adenocarcinoma Thomas G. Paulson,1,2 Carlo C. Maley,4,5 Xiaohong Li,2 Hongzhe Li,6 Carissa A. Sanchez,1,2 Dennis L. Chao,3 Robert D. Odze,7 Thomas L. Vaughan,2,8 Patricia L. Blount,1,2,10 and Brian J. Reid1,2,9,10 Abstract Purpose: Chromosomal instability, as assessed by many techniques, including DNA content aneuploidy, loss of heterozygosity, and comparative genomic hybridization, has consistently been reported to be common in cancer and rare in normal tissues. Recently, a panel of chromosome instability biomarkers, including loss of heterozygos- ity and DNA content, has been reported to identify patients at high and low risk of pro- gression from Barrett's esophagus (BE) to esophageal adenocarcinoma (EA), but required multiple platforms for implementation. Although chromosomal instability in- volving amplifications and deletions of chromosome regions have been observed in nearly all cancers, copy number alterations (CNA) in premalignant tissues have not been well characterized or evaluated in cohort studies as biomarkers of cancer risk. Experimental Design: We examined CNAs in 98 patients having either BE or EA using Bacterial Artificial Chromosome (BAC) array comparative genomic hybridization to char- acterize CNAs at different stages of progression ranging from early BE to advanced EA. Results: CNAs were rare in early stages (less than high-grade dysplasia) but were pro- gressively more frequent and larger in later stages (high-grade dysplasia and EA), in- cluding high-level amplifications. The number of CNAs correlated highly with DNA content aneuploidy. -

Alzheimer's Disease Is Associated with Reduced Expression of Energy
Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons Winnie S. Liang*†, Eric M. Reiman*†‡§, Jon Valla†¶, Travis Dunckley*†, Thomas G. Beach†ʈ, Andrew Grover†ʈ, Tracey L. Niedzielko†¶, Lonnie E. Schneider†¶, Diego Mastroeni†ʈ, Richard Caselli†**, Walter Kukull††, John C. Morris‡‡, Christine M. Hulette§§, Donald Schmechel§§, Joseph Rogers†ʈ, and Dietrich A. Stephan*†¶¶ *Neurogenomics Division, Translational Genomics Research Institute, 445 North Fifth Street, Phoenix, AZ 85004; †Arizona Alzheimer’s Consortium, 901 East Willetta Street, Phoenix, AZ 85006; ‡Banner Alzheimer’s Institute, 901 East Willetta Street Phoenix, AZ 85006; §Department of Psychiatry and Evelyn F. McKnight Brain Institute, University of Arizona, 1501 North Campbell Avenue, Tucson, AZ 85724; ¶Barrow Neurological Institute, 350 West Thomas Road, Phoenix, AZ 85013; ʈSun Health Research Institute, 10515 West Santa Fe Drive, Sun City, AZ 85351; **Department of Neurology, Mayo Clinic, 13400 East Shea Boulevard, Scottsdale, AZ 85259; ††National Alzheimer’s Coordinating Center, 4311 11th Avenue NE, No. 300, Seattle, WA 98105; ‡‡Washington University Alzheimer’s Disease Research Center, Washington University School of Medicine, 4488 Forest Park Avenue, Suite 101, St. Louis, MO 63108; and §§Bryan Alzheimer’s Disease Research Center, Duke University Medical Center, 2200 West Main Street, Suite A200, Durham, NC 27705 Edited by Marcus E. Raichle, Washington University School of Medicine, St. Louis, MO, and approved January 15, 2008 (received for review September 28, 2007) Alzheimer’s disease (AD) is associated with regional reductions in of genetic risk for AD) and were progressive in late-middle-aged fluorodeoxyglucose positron emission tomography (FDG PET) mea- persons (19). -
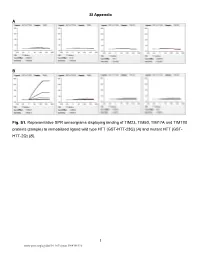
Sample) to Immobilized Ligand Wild Type HTT (GST-HTT-23Q) (A) and Mutant HTT (GST- HTT-2Q) (B)
SI Appendix A B Fig. S1. Representative SPR sensorgrams displaying binding of TIM23, TIM50, TIM17A and TIM17B proteins (sample) to immobilized ligand wild type HTT (GST-HTT-23Q) (A) and mutant HTT (GST- HTT-2Q) (B). 1 www.pnas.org/cgi/doi/10.1073/pnas.1904101116 Fig. S2. The level of mtCO1 protein is equal in equivalent number of mitochondria from ST-Hdh- Q7/Q7 and -Q111/Q111 cells. Representative immunoblot (A) and quantification (B) of mtCO1 level in FACS sorted mitochondria isolated from mito-eGFP transfected cells. Four million (4 MM) eGFP labeled mitochondria and 5 µg protein samples were immunoblotted for mtCO1, and nuclearly- encoded matrix aconitase 2 (ACO2) and outer membrane (SAM50) proteins. WCL – whole cell lysate, MITO – mitochondria, N.T. – not transfected, OMM – outer mitochondrial membrane. (n=3, data shown as mean+s.e.m. ** P<0.001, * P<0.05, t-Test). 2 A B 3 Fig. S3. Purity of mitochondria enriched fractions. (A) Representative immunoblot of Q7 and Q111 whole cell lysates (WCL), mitochondrial (Mito.) and cytosolic fractions (Cyto.) probed for mitochondrial markers (VDAC1, ATP5A, ACO2 and mtCO1) and typical mitochondrial fraction contaminants and contaminating organelles: cytosol (TUBA), endoplasmic reticulum (CALR), lysosomes (LAMP1), Golgi apparatus (RCAS1) and plasma membrane (CAV2). (B) Nonsynaptosomal (NS) and synaptosomal (SM) mitochondrial fractions isolated from fresh post- surgical cortex and frozen specimens of cortex and striatum (tissue blocks listed in Table 1) were tested for enrichment with mitochondrial markers and contamination with markers listed in (A) compared to whole brain homogenate (WB) and cytosolic fraction. 4 Fig. S4. Representative immunoblots of mitochondrial proteins from Q7 and Q111 cells (shown here to supplement Fig.