Application of Metabolic Flux and Transcriptome Analyses to Understanding the Physiology of Engineered Geobacillus Thermoglucosi
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Information
Supplementary Information Table S1. Pathway analysis of the 1246 dwf1-specific differentially expressed genes. Fold Change Fold Change Fold Change Gene ID Description (dwf1/WT) (XL-5/WT) (XL-6/WT) Carbohydrate Metabolism Glycolysis/Gluconeogenesis POPTR_0008s11770.1 Glucose-6-phosphate isomerase −1.7382 0.512146 0.168727 POPTR_0001s47210.1 Fructose-bisphosphate aldolase, class I 1.599591 0.044778 0.18237 POPTR_0011s05190.3 Probable phosphoglycerate mutase −2.11069 −0.34562 −0.9738 POPTR_0012s01140.1 Pyruvate kinase −1.25054 0.074697 −0.16016 POPTR_0016s12760.1 Pyruvate decarboxylase 2.664081 0.021062 0.371969 POPTR_0012s08010.1 Aldehyde dehydrogenase (NAD+) −1.41556 0.479957 −0.21366 POPTR_0014s13710.1 Acetyl-CoA synthetase −1.337 0.154552 −0.26532 POPTR_0017s11660.1 Aldose 1-epimerase 2.770518 0.016874 0.73016 POPTR_0010s11970.1 Phosphoglucomutase −1.25266 −0.35581 0.074064 POPTR_0012s14030.1 Phosphoglucomutase −1.15872 −0.68468 −0.93596 POPTR_0002s10850.1 Phosphoenolpyruvate carboxykinase (ATP) 1.489119 0.967284 0.821559 Citrate cycle (TCA cycle) 2-Oxoglutarate dehydrogenase E2 component POPTR_0014s15280.1 −1.63733 0.076435 0.170827 (dihydrolipoamide succinyltransferase) POPTR_0002s26120.1 Succinyl-CoA synthetase β subunit −1.29244 −0.38517 −0.3497 POPTR_0007s12750.1 Succinate dehydrogenase (ubiquinone) flavoprotein subunit −1.83751 0.519356 0.309149 POPTR_0002s10850.1 Phosphoenolpyruvate carboxykinase (ATP) 1.489119 0.967284 0.821559 Pentose phosphate pathway POPTR_0008s11770.1 Glucose-6-phosphate isomerase −1.7382 0.512146 0.168727 POPTR_0013s00660.1 Glucose-6-phosphate 1-dehydrogenase −1.26949 −0.18314 0.374822 POPTR_0015s00960.1 6-Phosphogluconolactonase 2.022223 0.168877 0.971431 POPTR_0010s11970.1 Phosphoglucomutase −1.25266 −0.35581 0.074064 POPTR_0012s14030.1 Phosphoglucomutase −1.15872 −0.68468 −0.93596 POPTR_0001s47210.1 Fructose-bisphosphate aldolase, class I 1.599591 0.044778 0.18237 S2 Table S1. -

Electronic Supplementary Information S10
Electronic Supplementary Material (ESI) for Metallomics. This journal is © The Royal Society of Chemistry 2019 Electronic Supplementary Information S10. Up and downregulated genes of ACR3 and TIP-ACR3 compared to control roots all exposed to 0.1 mM As III . HYBRIDIZATION 4: LIST OF UP-REGULATED GENES Fold Change Probe Set ID ([0.1 ACR3] vs [0.1-HR]) Blast2GO description Genbank Accessions C228_s_at 48.018467 N.tabacum cysteine-rich extensin-like protein-4 mRNA EB683071 C1359_at 32.31786 Proteinase inhibitor I3, Kunitz legume, Kunitz inhibitor ST1-like DW004832 EB430244_x_at 22.426174 unknow EB430244 C10896_at 21.463442 dir1 (defective in induced resistance 1) lipid binding JF275847.1 BP528597_at 21.445984 Mitochondrial DNA BP528597 C10933_x_at 20.347004 Solanum nigrum clone 82 organ-specific protein S2 (OS) EB443218 TT31_B05_s_at 17.260008 Proteinase inhibitor I3, Kunitz legume, Kunitz inhibitor ST1-like C3546_s_at 17.171844 fasciclin-like arabinogalactan protein 2 EB451563 C8455_at 16.855278 Defective in induced resistance 2 protein (DIR2) BP530866 C11687_at 16.618454 galactinol synthase EB432401 BP136836_s_at 15.364014 Nicotiana tabacum mitochondrial DNA BP136836 BP525701_at 15.137816 Nicotiana tabacum mitochondrial DNA BP525701 EB682942_at 14.943408 Hop-interacting protein THI101 EB682942 BP133164_at 14.644844 Nicotiana tabacum mitochondrial DNA BP133164 AY055111_at 14.570147 Nicotiana tabacum pathogenesis-related protein PR10a AY055111 TT08_C02_at 14.375496 BP526999_at 14.374481 Nicotiana tabacum mitochondrial DNA BP526999 CV017694_s_at -

Suberin Biosynthesis and Deposition in the Wound-Healing Potato (Solanum Tuberosum L.) Tuber Model
Western University Scholarship@Western Electronic Thesis and Dissertation Repository 12-4-2018 2:30 PM Suberin Biosynthesis and Deposition in the Wound-Healing Potato (Solanum tuberosum L.) Tuber Model Kathlyn Natalie Woolfson The University of Western Ontario Supervisor Bernards, Mark A. The University of Western Ontario Graduate Program in Biology A thesis submitted in partial fulfillment of the equirr ements for the degree in Doctor of Philosophy © Kathlyn Natalie Woolfson 2018 Follow this and additional works at: https://ir.lib.uwo.ca/etd Part of the Plant Biology Commons Recommended Citation Woolfson, Kathlyn Natalie, "Suberin Biosynthesis and Deposition in the Wound-Healing Potato (Solanum tuberosum L.) Tuber Model" (2018). Electronic Thesis and Dissertation Repository. 5935. https://ir.lib.uwo.ca/etd/5935 This Dissertation/Thesis is brought to you for free and open access by Scholarship@Western. It has been accepted for inclusion in Electronic Thesis and Dissertation Repository by an authorized administrator of Scholarship@Western. For more information, please contact [email protected]. Abstract Suberin is a heteropolymer comprising a cell wall-bound poly(phenolic) domain (SPPD) covalently linked to a poly(aliphatic) domain (SPAD) that is deposited between the cell wall and plasma membrane. Potato tuber skin contains suberin to protect against water loss and microbial infection. Wounding triggers suberin biosynthesis in usually non- suberized tuber parenchyma, providing a model system to study suberin production. Spatial and temporal coordination of SPPD and SPAD-related metabolism are required for suberization, as the former is produced soon after wounding, and the latter is synthesized later into wound-healing. Many steps involved in suberin biosynthesis remain uncharacterized, and the mechanism(s) that regulate and coordinate SPPD and SPAD production and assembly are not understood. -

The Prephenate Dehydrogenase Component of the Bifunctional T-Protein in Enteric Bacteria Can Utilize L-Arogenate
CORE Metadata, citation and similar papers at core.ac.uk Provided by Elsevier - Publisher Connector Volume 216, number 1, 133-139 FEB 04718 May 1987 The prephenate dehydrogenase component of the bifunctional T-protein in enteric bacteria can utilize L-arogenate Suhail Ahmad and Roy A. Jensen Department of Microbiology and Cell Science, McCarty Hall, IFAS, University of Florida, Gainesville, FL 32611, USA Received 3 February 1987; revised version received 19 March 1987 The prephenate dehydrogenase component of the bifunctional T-protein (chorismate mutase:prephenate de- hydrogenase) has been shown to utilize L-arogenate, a common precursor of phenylalanine and tyrosine in nature, as a substrate. Partially purified T-protein from Klebsiellapneumoniae and from Escherichia coli strains K12, B, C and W was used to demonstrate the utilization of L-arogenate as an alternative substrate for prephenate in the presence of nlcotinamide adenine dinucleotide as cofactor. The formation of L-tyro- sine from L-arogenate by the T-protein dehydrogenase was confirmed by high-performance liquid chroma- tography. As expected of a common catalytic site, dehydrogenase activity with either prephenate or L-aroge- nate was highly sensitive to inhibition by L-tyrosine. Tyrosine synthesis; T-protein; Enteric bacteria; Regulatory enzyme 1. INTRODUCTION specifying chorismate mutase [2]. Cyclohexadienyl dehydrogenase and cyclohexadienyl dehydratase Escherichia coli and Klebsiella pneumoniae, (often referred to as arogenate dehydrogenase and closely related enteric bacteria, possess a pair of arogenate dehydratase in earlier papers) are able to bifunctional proteins that compete for chorismate accept either prephenate or L-arogenate as as initial substrate molecules in reactions leading to substrates. -

Modeling and Computational Prediction of Metabolic Channelling
MODELING AND COMPUTATIONAL PREDICTION OF METABOLIC CHANNELLING by Christopher Morran Sanford A thesis submitted in conformity with the requirements for the degree of Master of Science Graduate Department of Molecular Genetics University of Toronto © Copyright by Christopher Morran Sanford 2009 Abstract MODELING AND COMPUTATIONAL PREDICTION OF METABOLIC CHANNELLING Master of Science 2009 Christopher Morran Sanford Graduate Department of Molecular Genetics University of Toronto Metabolic channelling occurs when two enzymes that act on a common substrate pass that intermediate directly from one active site to the next without allowing it to diffuse into the surrounding aqueous medium. In this study, properties of channelling are investigated through the use of computational models and cell simulation tools. The effects of enzyme kinetics and thermodynamics on channelling are explored with the emphasis on validating the hypothesized roles of metabolic channelling in living cells. These simulations identify situations in which channelling can induce acceleration of reaction velocities and reduction in the free concentration of intermediate metabolites. Databases of biological information, including metabolic, thermodynamic, toxicity, inhibitory, gene fusion and physical protein interaction data are used to predict examples of potentially channelled enzyme pairs. The predictions are used both to support the hypothesized evolutionary motivations for channelling, and to propose potential enzyme interactions that may be worthy of future investigation. ii Acknowledgements I wish to thank my supervisor Dr. John Parkinson for the guidance he has provided during my time spent in his lab, as well as for his extensive help in the writing of this thesis. I am grateful for the advice of my committee members, Prof. -

Plastid-Localized Amino Acid Biosynthetic Pathways of Plantae Are Predominantly Composed of Non-Cyanobacterial Enzymes
Plastid-localized amino acid biosynthetic pathways of Plantae are predominantly SUBJECT AREAS: MOLECULAR EVOLUTION composed of non-cyanobacterial PHYLOGENETICS PLANT EVOLUTION enzymes PHYLOGENY Adrian Reyes-Prieto1* & Ahmed Moustafa2* Received 1 26 September 2012 Canadian Institute for Advanced Research and Department of Biology, University of New Brunswick, Fredericton, Canada, 2Department of Biology and Biotechnology Graduate Program, American University in Cairo, Egypt. Accepted 27 November 2012 Studies of photosynthetic eukaryotes have revealed that the evolution of plastids from cyanobacteria Published involved the recruitment of non-cyanobacterial proteins. Our phylogenetic survey of .100 Arabidopsis 11 December 2012 nuclear-encoded plastid enzymes involved in amino acid biosynthesis identified only 21 unambiguous cyanobacterial-derived proteins. Some of the several non-cyanobacterial plastid enzymes have a shared phylogenetic origin in the three Plantae lineages. We hypothesize that during the evolution of plastids some enzymes encoded in the host nuclear genome were mistargeted into the plastid. Then, the activity of those Correspondence and foreign enzymes was sustained by both the plastid metabolites and interactions with the native requests for materials cyanobacterial enzymes. Some of the novel enzymatic activities were favored by selective compartmentation should be addressed to of additional complementary enzymes. The mosaic phylogenetic composition of the plastid amino acid A.R.-P. ([email protected]) biosynthetic pathways and the reduced number of plastid-encoded proteins of non-cyanobacterial origin suggest that enzyme recruitment underlies the recompartmentation of metabolic routes during the evolution of plastids. * Equal contribution made by these authors. rimary plastids of plants and algae are the evolutionary outcome of an endosymbiotic association between eukaryotes and cyanobacteria1. -

Role of Cytosolic, Tyrosine-Insensitive Prephenate Dehydrogenase in Medicago Truncatula
bioRxiv preprint doi: https://doi.org/10.1101/768317; this version posted September 13, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 2 Title of article: Role of Cytosolic, Tyrosine-Insensitive Prephenate Dehydrogenase in 3 Medicago truncatula 4 5 6 Authors: Craig A. Schenck1,2, Josh Westphal1, Dhileepkumar Jayaraman3, Kevin Garcia3,4, 7 Jiangqi Wen5, Kirankumar S. Mysore5 Jean-Michel Ané3,6, Lloyd W. Sumner7,8 and Hiroshi A. 8 Maeda1,* 9 10 11 Affiliations: 1Department of Botany, University of Wisconsin-Madison, Madison, WI 53706 12 2 Current address: Department of Biochemistry and Molecular Biology, Michigan State 13 University, East Lansing, MI 48824 14 3 Department of Bacteriology, University of Wisconsin-Madison, Madison, WI 53706 15 4 Department of Crop and Soil Sciences, North Carolina State University, Raleigh, NC 27695 16 5 Noble Research Institute, LLC., Ardmore, OK, USA 17 6 Department of Agronomy, University of Wisconsin-Madison, Madison, WI 53706 18 7 Department of Biochemistry, University of Missouri, Columbia, MO 65211 19 8 Metabolomics and Bond Life Sciences Centers, University of Missouri, Colubmia, MO 65211 20 21 22 23 *Corresponding author: Hiroshi A. Maeda ([email protected]) 24 25 26 1 bioRxiv preprint doi: https://doi.org/10.1101/768317; this version posted September 13, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 27 ABSTRACT 28 L-Tyrosine (Tyr) is an aromatic amino acid synthesized de novo in plants and microbes 29 downstream of the shikimate pathway. -

All Enzymes in BRENDA™ the Comprehensive Enzyme Information System
All enzymes in BRENDA™ The Comprehensive Enzyme Information System http://www.brenda-enzymes.org/index.php4?page=information/all_enzymes.php4 1.1.1.1 alcohol dehydrogenase 1.1.1.B1 D-arabitol-phosphate dehydrogenase 1.1.1.2 alcohol dehydrogenase (NADP+) 1.1.1.B3 (S)-specific secondary alcohol dehydrogenase 1.1.1.3 homoserine dehydrogenase 1.1.1.B4 (R)-specific secondary alcohol dehydrogenase 1.1.1.4 (R,R)-butanediol dehydrogenase 1.1.1.5 acetoin dehydrogenase 1.1.1.B5 NADP-retinol dehydrogenase 1.1.1.6 glycerol dehydrogenase 1.1.1.7 propanediol-phosphate dehydrogenase 1.1.1.8 glycerol-3-phosphate dehydrogenase (NAD+) 1.1.1.9 D-xylulose reductase 1.1.1.10 L-xylulose reductase 1.1.1.11 D-arabinitol 4-dehydrogenase 1.1.1.12 L-arabinitol 4-dehydrogenase 1.1.1.13 L-arabinitol 2-dehydrogenase 1.1.1.14 L-iditol 2-dehydrogenase 1.1.1.15 D-iditol 2-dehydrogenase 1.1.1.16 galactitol 2-dehydrogenase 1.1.1.17 mannitol-1-phosphate 5-dehydrogenase 1.1.1.18 inositol 2-dehydrogenase 1.1.1.19 glucuronate reductase 1.1.1.20 glucuronolactone reductase 1.1.1.21 aldehyde reductase 1.1.1.22 UDP-glucose 6-dehydrogenase 1.1.1.23 histidinol dehydrogenase 1.1.1.24 quinate dehydrogenase 1.1.1.25 shikimate dehydrogenase 1.1.1.26 glyoxylate reductase 1.1.1.27 L-lactate dehydrogenase 1.1.1.28 D-lactate dehydrogenase 1.1.1.29 glycerate dehydrogenase 1.1.1.30 3-hydroxybutyrate dehydrogenase 1.1.1.31 3-hydroxyisobutyrate dehydrogenase 1.1.1.32 mevaldate reductase 1.1.1.33 mevaldate reductase (NADPH) 1.1.1.34 hydroxymethylglutaryl-CoA reductase (NADPH) 1.1.1.35 3-hydroxyacyl-CoA -

Vhe Aromatic Amino Acid Pathway Branches at L-Arogenate In,Uglena Gracilisl7j3, GRAHAM S
MOLECULAR AND CELLULAR BIOLOGY, May 1981, p. 426-438 Vol. 1, No. 5 0270-7306/81/050426-13$2.00/0 )Vhe Aromatic Amino Acid Pathway Branches at L-Arogenate in,uglena gracilisL7j3, GRAHAM S. BYNG, ROBERT J.JWHITAKER, CHARLES L.(SHAPIRO, AND ROY A.4:NSEN* Center for Soma c-Cell Genetics and Biochemistry, Department of Biological Sciences, State University of New York at Binghamton, Binghamton, New York 13901 Received 5 December 1980/Accepted 6 March 1981 The recently characterized amino acid L-arogenate (Zamir et al., J. Am. Chem. Soc. 102:4499-4504, 1980) may be a precursor of either L-phenylalanine or L- tyrosine in nature. Euglena gracilis is the first example of an organism that uses L-arogenate as the sole precursor of both L-tyrosine and L-phenylalanine, thereby creating a pathway in which L-arogenate rather than prephenate becomes the metabolic branch point. E. gracilis ATCC 12796 was cultured in the light under myxotrophic conditions and harvested in late exponential phase before extract preparation for enzymological assays. Arogenate dehydrogenase was dependent upon nicotinamide adenine dinucleotide phosphate for activity. L-Tyrosine in- hibited activity effectively with kinetics that were competitive with respect to L- arogenate and noncompetitive with respect to nicotinamide adenine dinucleotide phosphate. The possible inhibition of arogenate dehydratase by L-phenylalanine has not yet been determined. Beyond the latter uncertainty, the overall regulation of aromatic biosynthesis was studied through the characterization of 3-deoxy-D- arabino-heptulosonate 7-phosphate synthase and chorismate mutase. 3-Deoxy- D-arabino-heptulosonate 7-phosphate synthase was subject to noncompetitive inhibition by L-tyrosine with respect to either of the two substrates. -
(12) United States Patent (10) Patent No.: US 8,883,464 B2 Lynch Et Al
USOO8883464B2 (12) United States Patent (10) Patent No.: US 8,883,464 B2 Lynch et al. (45) Date of Patent: Nov. 11, 2014 (54) METHODS FOR PRODUCING (56) References Cited 3-HYDROXYPROPIONCACID AND OTHER U.S. PATENT DOCUMENTS PRODUCTS M 2.408,889 A 10, 1946 Short (75) Inventors: Michael D. Lynch, Boulder, CO (US); 2.464,768 A 3, 1949 Redmon et al. Ryan T. Gill, Denver, CO (US); Tanya 2.469,701 A 5/1949 Redmon E. W. Lipscomb, Boulder, CO (US) 3,904,685.2,798,053 A 9/19757, 1957 ShahidiBrown et et al. al. (73) Assignees: OPX Biotechnologies, Inc., Boulder, 4,029,5773,915,921 A 10/19756, 1977 Schlatzer,E. Jr. s al. CO (US); The Regents of the 4,268,641 A 5/1981 Koenig et al. University of Colorado, a Body 1945. A 13. Mister et al. Corporate, Denver, CO (US) 4,666,983.I - J. A 5/1987 Tsubakimoto et al. 4,685,915 A 8, 1987 Hasse et al. (*) Notice: Subject to any disclaimer, the term of this 4,708,997 A 1 1/1987 Stanley, Jr. et al. patent is extended or adjusted under 35 3. A s 3. Its et al. U.S.C. 154(b) by 0 days. 4,952.505. A 8/1990 Chomelir et al. 4,985,518 A 1/1991 Alexander et al. (21) Appl. No.: 13/498,468 5,009,653 A 4/1991 Osborn, III 5,093,472 A 3, 1992 Bresciani (22) PCT Filed: Sep. 27, 2010 5,135,677 A 8/1992 Yamaguchi et al. -

Springer Handbook of Enzymes
Dietmar Schomburg Ida Schomburg (Eds.) Springer Handbook of Enzymes Alphabetical Name Index 1 23 © Springer-Verlag Berlin Heidelberg New York 2010 This work is subject to copyright. All rights reserved, whether in whole or part of the material con- cerned, specifically the right of translation, printing and reprinting, reproduction and storage in data- bases. The publisher cannot assume any legal responsibility for given data. Commercial distribution is only permitted with the publishers written consent. Springer Handbook of Enzymes, Vols. 1–39 + Supplements 1–7, Name Index 2.4.1.60 abequosyltransferase, Vol. 31, p. 468 2.7.1.157 N-acetylgalactosamine kinase, Vol. S2, p. 268 4.2.3.18 abietadiene synthase, Vol. S7,p.276 3.1.6.12 N-acetylgalactosamine-4-sulfatase, Vol. 11, p. 300 1.14.13.93 (+)-abscisic acid 8’-hydroxylase, Vol. S1, p. 602 3.1.6.4 N-acetylgalactosamine-6-sulfatase, Vol. 11, p. 267 1.2.3.14 abscisic-aldehyde oxidase, Vol. S1, p. 176 3.2.1.49 a-N-acetylgalactosaminidase, Vol. 13,p.10 1.2.1.10 acetaldehyde dehydrogenase (acetylating), Vol. 20, 3.2.1.53 b-N-acetylgalactosaminidase, Vol. 13,p.91 p. 115 2.4.99.3 a-N-acetylgalactosaminide a-2,6-sialyltransferase, 3.5.1.63 4-acetamidobutyrate deacetylase, Vol. 14,p.528 Vol. 33,p.335 3.5.1.51 4-acetamidobutyryl-CoA deacetylase, Vol. 14, 2.4.1.147 acetylgalactosaminyl-O-glycosyl-glycoprotein b- p. 482 1,3-N-acetylglucosaminyltransferase, Vol. 32, 3.5.1.29 2-(acetamidomethylene)succinate hydrolase, p. 287 Vol. -
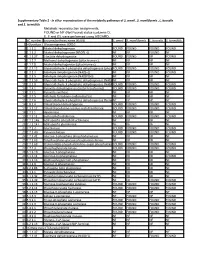
Supplementary Table 2 - in Silico Reconstruction of the Metabolic Pathways of S
Supplementary Table 2 - In silico reconstruction of the metabolic pathways of S. amnii , S. moniliformis , L. buccalis and S. termiditis Metabolic reconstruction assignments, FOUND or NF (Not Found) status (columns D, E, F and G), were performed using ASGARD, EC number Enzyme/pathway name (KEGG) S. amnii S. moniliformis L. buccalis S. termiditis 1 >Glycolysis / Gluconeogenesis 00010 2 1.1.1.1 Alcohol dehydrogenase. FOUND FOUND FOUND FOUND 3 1.1.1.2 Alcohol dehydrogenase (NADP(+)). NF NF FOUND NF 4 1.1.1.27 L-lactate dehydrogenase. FOUND FOUND NF FOUND 5 1.1.2.7 Methanol dehydrogenase (cytochrome c). NF NF NF NF 6 1.1.2.8 Alcohol dehydrogenase (cytochrome c). NF NF NF NF 7 1.2.1.12 Glyceraldehyde-3-phosphate dehydrogenase (phosphorylating).FOUND FOUND FOUND FOUND 8 1.2.1.3 Aldehyde dehydrogenase (NAD(+)). NF NF FOUND FOUND 9 1.2.1.5 Aldehyde dehydrogenase (NAD(P)(+)). NF NF NF NF 10 1.2.1.59 Glyceraldehyde-3-phosphate dehydrogenase (NAD(P)(+))NF (phosphorylating).NF NF NF 11 1.2.1.9 Glyceraldehyde-3-phosphate dehydrogenase (NADP(+)).FOUND FOUND FOUND FOUND 12 1.2.4.1 Pyruvate dehydrogenase (acetyl-transferring). FOUND FOUND FOUND FOUND 13 1.2.7.1 Pyruvate synthase. NF NF NF NF 14 1.2.7.5 Aldehyde ferredoxin oxidoreductase. NF NF NF NF 15 1.2.7.6 Glyceraldehyde-3-phosphate dehydrogenase (ferredoxin).NF NF NF NF 16 1.8.1.4 Dihydrolipoyl dehydrogenase. FOUND FOUND FOUND FOUND 17 2.3.1.12 Dihydrolipoyllysine-residue acetyltransferase. FOUND FOUND FOUND FOUND 18 2.7.1.1 Hexokinase.