Genome-Wide Association Studies
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Whole Genome Sequencing Identifies Novel Structural Variant in a Large Indian Family Affected with X - Linked Agammaglobulinemia
medRxiv preprint doi: https://doi.org/10.1101/2020.10.05.20200949; this version posted October 6, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-ND 4.0 International license . Whole Genome Sequencing identifies novel structural variant in a large Indian family affected with X - linked agammaglobulinemia 1,2,# 3,5,#,$ 3,4 3 Abhinav Jain , Geeta Madathil Govindaraj , Athulya Edavazhippurath , Nabeel Faisal , Rahul C 1 1,2 6 3 1 1 Bhoyar , Vishu Gupta , Ramya Uppuluri , Shiny Padinjare Manakkad , Atul Kashyap , Anoop Kumar , 1,2 1,2 7 7 3 Mohit Kumar Divakar , Mohamed Imran , Sneha Sawant , Aparna Dalvi , Krishnan Chakyar , 7 6 1,2,$ 1,2,$ Manisha Madkaikar , Revathi Raj , Sridhar Sivasubbu , Vinod Scaria 1 CSIR-Institute of Genomics and Integrative Biology, Mathura Road, New Delhi, Delhi, India 2 Academy of Scientific and Innovative Research (AcSIR), Mathura Road, New Delhi, Delhi, India 3 Department of Pediatrics, Government Medical College Kozhikode, Kozhikode, Kerala, India 4 Multidisciplinary Research Unit, Government College Kozhikode, Kozhikode, Kerala, India 5 FPID Regional Diagnostic Centre, Government Medical College Kozhikode, Kozhikode, Kerala India 6 Department of Pediatric Hematology, Oncology, Blood and Marrow Transplantation, Apollo Hospitals, 320, Padma complex, Anna salai, Teynampet, Chennai, Tamil Nadu, India 7 Department of Pediatric Immunology and Leukocyte Biology, ICMR-National Institute of Immunohaematology, KEM Hospital, Parel, Mumbai, Maharashtra, India. # Joint first authors $ Corresponding author Vinod Scaria: [email protected] Sridhar Sivasubbu: [email protected] Geeta Madathil Govindaraj: [email protected] NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. -

The Precision Medicine Issue Medicine Is Becoming Hyper
The precision medicine issue Vol. 121 Nov/Dec $9.99 USD No. 6 2018 $10.99 CAD $2 million would save her life. The precision medicine issue Could you pay? Should you? Medicine is becoming hyper-personalized, hyper-accurate ... and hyper-unequal. p.38 The AIs taking over Curing cancer How to plan your from doctors with customized digital afterlife vaccines p.22 p.46 p.76 ND18_cover.indd 1 10/3/18 4:16 PM Artificial intelligence is here. Own what happens next. ND18 ETD19 Spread 16x10.875 D3.indd 2 9/28/18 10:59 AM March 25–26, 2019 St. egis Hotel San Francisco, CA Meet the people leading the next wave of intelligent technologies. Andrew Kathy Daphne Sergey Harry Anagnost Gong Koller Levine Shum President and Cofounder and Founder and Assistant Executive Vice CEO, Autodesk CEO, WafaGames CEO, Insitro Professor, EECS, President, UC Berkeley Microsoft Artifi cial Intelligence and Research Group EmTechDigital.com/2019 ND18 ETD19 Spread 16x10.875 D3.indd 3 9/28/18 10:59 AM 02 From the editor It’s been nearly two decades since the help meet the ballooning healthcare first human genome was sequenced. needs of an aging population. That achievement opened up the prom The problem? As medicine gets ise of drugs and treatments perfectly more personalized, it risks getting more tailored to each person’s DNA. But per unequal. Our cover story is Regalado’s sonalized or “precision” medicine has gripping and troubling account page always felt a bit like flying cars, sexbots, 38 of the parents who raise millions of or labgrown meatone of those things dollars to finance genetherapy cures for we’re perpetually being promised but their children’s ultrarare diseases. -

Exome Sequencing and Functional Validation in Zebrafish Identify
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector REPORT Exome Sequencing and Functional Validation in Zebrafish Identify GTDC2 Mutations as a Cause of Walker-Warburg Syndrome M. Chiara Manzini,1,2 Dimira E. Tambunan,1,2 R. Sean Hill,1,2 Tim W. Yu,1,2 Thomas M. Maynard,3 Erin L. Heinzen,4 Kevin V. Shianna,4 Christine R. Stevens,5 Jennifer N. Partlow,1,2 Brenda J. Barry,1,2 Jacqueline Rodriguez,1,2 Vandana A. Gupta,1,6 Abdel-Karim Al-Qudah,7 Wafaa M. Eyaid,8 Jan M. Friedman,9,10 Mustafa A. Salih,11 Robin Clark,12 Isabella Moroni,13 Marina Mora,14 Alan H. Beggs,1,6 Stacey B. Gabriel,5 and Christopher A. Walsh1,2,5,* Whole-exome sequencing (WES), which analyzes the coding sequence of most annotated genes in the human genome, is an ideal approach to studying fully penetrant autosomal-recessive diseases, and it has been very powerful in identifying disease-causing muta- tions even when enrollment of affected individuals is limited by reduced survival. In this study, we combined WES with homozygosity analysis of consanguineous pedigrees, which are informative even when a single affected individual is available, to identify genetic mutations responsible for Walker-Warburg syndrome (WWS), a genetically heterogeneous autosomal-recessive disorder that severely affects the development of the brain, eyes, and muscle. Mutations in seven genes are known to cause WWS and explain 50%–60% of cases, but multiple additional genes are expected to be mutated because unexplained cases show suggestive linkage to diverse loci. -

Genomic Approaches for Understanding the Genetics of Complex Disease
Downloaded from genome.cshlp.org on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press Perspective Genomic approaches for understanding the genetics of complex disease William L. Lowe Jr.1 and Timothy E. Reddy2,3 1Division of Endocrinology, Metabolism and Molecular Medicine, Department of Medicine, Northwestern University Feinberg School of Medicine, Chicago, Illinois 60611, USA; 2Department of Biostatistics and Bioinformatics, Duke University Medical School, Durham, North Carolina 27708, USA; 3Center for Genomic and Computational Biology, Duke University Medical School, Durham, North Carolina 27708, USA There are thousands of known associations between genetic variants and complex human phenotypes, and the rate of novel discoveries is rapidly increasing. Translating those associations into knowledge of disease mechanisms remains a fundamental challenge because the associated variants are overwhelmingly in noncoding regions of the genome where we have few guiding principles to predict their function. Intersecting the compendium of identified genetic associations with maps of regulatory activity across the human genome has revealed that phenotype-associated variants are highly enriched in candidate regula- tory elements. Allele-specific analyses of gene regulation can further prioritize variants that likely have a functional effect on disease mechanisms; and emerging high-throughput assays to quantify the activity of candidate regulatory elements are a promising next step in that direction. Together, these technologies have created the ability to systematically and empirically test hypotheses about the function of noncoding variants and haplotypes at the scale needed for comprehensive and system- atic follow-up of genetic association studies. Major coordinated efforts to quantify regulatory mechanisms across genetically diverse populations in increasingly realistic cell models would be highly beneficial to realize that potential. -

QTL Analysis Reveals Genomic Variants Linked to High-Temperature
Wang et al. Biotechnol Biofuels (2019) 12:59 https://doi.org/10.1186/s13068-019-1398-7 Biotechnology for Biofuels RESEARCH Open Access QTL analysis reveals genomic variants linked to high-temperature fermentation performance in the industrial yeast Zhen Wang1,2†, Qi Qi1,2†, Yuping Lin1*, Yufeng Guo1, Yanfang Liu1,2 and Qinhong Wang1* Abstract Background: High-temperature fermentation is desirable for the industrial production of ethanol, which requires thermotolerant yeast strains. However, yeast thermotolerance is a complicated quantitative trait. The understanding of genetic basis behind high-temperature fermentation performance is still limited. Quantitative trait locus (QTL) map- ping by pooled-segregant whole genome sequencing has been proved to be a powerful and reliable approach to identify the loci, genes and single nucleotide polymorphism (SNP) variants linked to quantitative traits of yeast. Results: One superior thermotolerant industrial strain and one inferior thermosensitive natural strain with distinct high-temperature fermentation performances were screened from 124 Saccharomyces cerevisiae strains as parent strains for crossing and segregant isolation. Based on QTL mapping by pooled-segregant whole genome sequencing as well as the subsequent reciprocal hemizygosity analysis (RHA) and allele replacement analysis, we identifed and validated total eight causative genes in four QTLs that linked to high-temperature fermentation of yeast. Interestingly, loss of heterozygosity in fve of the eight causative genes including RXT2, ECM24, CSC1, IRA2 and AVO1 exhibited posi- tive efects on high-temperature fermentation. Principal component analysis (PCA) of high-temperature fermentation data from all the RHA and allele replacement strains of those eight genes distinguished three superior parent alleles including VPS34, VID24 and DAP1 to be greatly benefcial to high-temperature fermentation in contrast to their inferior parent alleles. -

A Comprehensive Database of Putative Human Microrna Target Site
bioRxiv preprint doi: https://doi.org/10.1101/554485; this version posted May 26, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. dbMTS: a comprehensive database of putative human microRNA target site SNVs and their functional predictions Chang Li1,2, Michael D. Swartz3, Bing Yu1, Yongsheng Bai4, Xiaoming Liu1,2* 1Human Genetics Center and Department of Epidemiology, Human Genetics and Environmental Sciences, School of Public Health, The University of Texas Health Science Center at Houston, Houston, TX 2USF Genomics, College of Public Health, University of South Florida, Tampa, FL 3Department of Biostatistics, School of Public Health, The University of Texas Health Science Center at Houston, Houston, TX 4Department of Internal Medicine, University of Michigan, Ann Arbor, MI *Correspondence: Xiaoming Liu, [email protected] Funding information: This work is supported partially by NIH funding 1UM1HG008898 and partially by the startup funding for XL from the University of South Florida. bioRxiv preprint doi: https://doi.org/10.1101/554485; this version posted May 26, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract microRNAs (miRNAs) are short non-coding RNAs that can repress the expression of protein coding messenger RNAs (mRNAs) by binding to the 3’UTR of the target. -

Interleukin 10 Mutant Zebrafish Have an Enhanced Interferon
www.nature.com/scientificreports OPEN Interleukin 10 mutant zebrafsh have an enhanced interferon gamma response and improved Received: 22 August 2017 Accepted: 20 June 2018 survival against a Mycobacterium Published: xx xx xxxx marinum infection Sanna-Kaisa E. Harjula1, Markus J. T. Ojanen1,2, Sinja Taavitsainen3, Matti Nykter3 & Mika Rämet1,4,5,6 Tuberculosis ranks as one of the world’s deadliest infectious diseases causing more than a million casualties annually. IL10 inhibits the function of Th1 type cells, and IL10 defciency has been associated with an improved resistance against Mycobacterium tuberculosis infection in a mouse model. Here, we utilized M. marinum infection in the zebrafsh (Danio rerio) as a model for studying Il10 in the host response against mycobacteria. Unchallenged, nonsense il10e46/e46 mutant zebrafsh were fertile and phenotypically normal. Following a chronic mycobacterial infection, il10e46/e46 mutants showed enhanced survival compared to the controls. This was associated with an increased expression of the Th cell marker cd4-1 and a shift towards a Th1 type immune response, which was demonstrated by the upregulated expression of tbx21 and ifng1, as well as the down-regulation of gata3. In addition, at 8 weeks post infection il10e46/e46 mutant zebrafsh had reduced expression levels of proinfammatory cytokines tnf and il1b, presumably indicating slower progress of the infection. Altogether, our data show that Il10 can weaken the immune defense against M. marinum infection in zebrafsh by restricting ifng1 response. Importantly, our fndings support the relevance of M. marinum infection in zebrafsh as a model for tuberculosis. Annually more than 10 million new tuberculosis cases are estimated to emerge, leading to over a million casualties1. -

Daniel J. Benjamin (As of 10/4/2019)
Daniel J. Benjamin (as of 10/4/2019) Contact Information University of Southern California Center for Economic and Social Research Dauterive Hall 635 Downey Way, Suite 301 Los Angeles, CA 90089-3332 Phone: 617-548-8948 Professional Experience: Professor (Research) of Economics, CESR, USC, 2019-present Associate Professor (Research) of Economics, CESR, USC, 2015-2019 Visiting Associate Professor (Research) of Economics, CESR, USC, 2014-2015 Associate Professor (with tenure), Economics Department, Cornell University, 2013-2015 Assistant Professor, Economics Department, Cornell University, 2007-2013 Visiting Research Fellow, National Bureau of Economic Research (NBER), 2011-2012 Research Associate, NBER, 2013-present Faculty Research Fellow, NBER, 2009-2013 Research Fellow, Research Center for Group Dynamics, Institute for Social Research, 2006-2017 Research Fellow, Population Studies Center, Institute for Social Research, 2006-2007 Graduate Studies: Ph.D., Economics, Harvard University, 2006 M.Sc., Mathematical Economics, London School of Economics, 2000 A.M., Statistics, Harvard University, 1999 Undergraduate Studies: A.B., Economics, Harvard University, summa cum laude, prize for best economics student, 1999 Honors, Scholarships, and Fellowships: 2013 Norwegian School of Economics Sandmo Junior Fellowship (prize for a “promising young economist”) 2012-2013 Cornell’s Institute for Social Science (ISS) Faculty Fellowship 2009-2012 ISS Theme Project Member: Judgment, Decision Making, and Social Behavior 2011-2012 NBER Visiting Fellowship -

Comparison of Structural and Short Variants Detectedby Linked-Read
cancers Article Comparison of Structural and Short Variants Detected by Linked-Read and Whole-Exome Sequencing in Multiple Myeloma Ashwini Kumar 1,2 , Sadiksha Adhikari 1,2, Matti Kankainen 2,3,4,5 and Caroline A. Heckman 1,2,* 1 Institute for Molecular Medicine Finland-FIMM, HiLIFE-Helsinki Institute of Life Science, iCAN Digital Cancer Medicine Flagship, University of Helsinki, Tukholmankatu 8, 00290 Helsinki, Finland; ashwini.kumar@helsinki.fi (A.K.); sadiksha.adhikari@helsinki.fi (S.A.) 2 iCAN Digital Precision Cancer Medicine, University of Helsinki, 00014 Helsinki, Finland; matti.kankainen@helsinki.fi 3 Medical and Clinical Genetics, University of Helsinki, Helsinki University Hospital, 00029 Helsinki, Finland 4 Translational Immunology Research Program and Department of Clinical Chemistry, University of Helsinki, 00290 Helsinki, Finland 5 Hematology Research Unit Helsinki, Department of Hematology, Helsinki University Hospital Comprehensive Cancer Center, 00290 Helsinki, Finland * Correspondence: caroline.heckman@helsinki.fi; Tel.: +358-29-412-5769 Simple Summary: The wide variety of next-generation sequencing technologies requires thorough evaluation and understanding of their advantages and shortcomings of these different approaches prior to their implementation in a precision medicine setting. Here, we compared the performance of two DNA sequencing methods, whole-exome and linked-read exome sequencing, to detect large Citation: Kumar, A.; Adhikari, S.; structural variants (SVs) and short variants in eight multiple myeloma (MM) patient cases. For three Kankainen, M.; Heckman, C.A. patient cases, matched tumor-normal samples were sequenced with both methods to compare somatic Comparison of Structural and Short SVs and short variants. The methods’ clinical relevance was also evaluated, and their sensitivity Variants Detected by Linked-Read and specificity to detect MM-specific cytogenetic alterations and other short variants were measured. -

Variant Annotation
Variant annotation Theorical part V1.1 10/10/18 Ready for annotating variants!! Variant annotation and prioritization Annotation Databases Eilbeck, Karen & Quinlan, Aaron & Yandell, Mark. (2017). Settling the score: variant prioritization and Mendelian disease. Nature Reviews Genetics. 10.1038/nrg.2017.52. Genomic data repositories - 1000 Genomes The 1000 Genomes Project (abbreviated as 1KGP), launched in January 2008, was an international research effort to establish by far the most detailed catalogue of human genetic variation. - ESP (NHLBI Exome Sequencing Project) Exists in 3 flavours : evs annotation data was generated from approximately 2500 exomes, evs_5400 from approximately 5400 exomes and the last one, evs_6500 from approximately 6500 exomes - ExAC (Exome Aggregation Consortium) Coalition of investigators seeking to aggregate and harmonize exome sequencing data from a variety of large-scale sequencing projects, and to make summary data available for the wider scientific community. - gnomAD (Genome Aggregation Database) Developed by an international coalition of investigators, with the goal of aggregating and harmonizing both exome and genome sequencing data from a wide variety of large-scale sequencing projects. - FREX (THE FRENCH EXOME) A REFERENCE PANEL OF EXOMES FROM FRENCH REGIONS Annotation Databases Eilbeck, Karen & Quinlan, Aaron & Yandell, Mark. (2017). Settling the score: variant prioritization and Mendelian disease. Nature Reviews Genetics. 10.1038/nrg.2017.52. Databases of variant-disease and gene-disease associations -

1 Mapping Challenging Mutations by Whole-Genome Sequencing Harold
bioRxiv preprint doi: https://doi.org/10.1101/036046; this version posted January 6, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Mapping challenging mutations by whole-genome sequencing Harold E. Smith, Amy S. Fabritius, Aimee Jaramillo-Lambert, and Andy Golden National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892 1 bioRxiv preprint doi: https://doi.org/10.1101/036046; this version posted January 6, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Running title: Challenging mutations by WGS Keywords: Forward genetics, variant detection, complex alleles, SNP mapping Corresponding author: Harold E. Smith 8 Center Drive Bethesda, MD 20892 301-594-6554 [email protected] 2 bioRxiv preprint doi: https://doi.org/10.1101/036046; this version posted January 6, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. ABSTRACT Whole-genome sequencing provides a rapid and powerful method for identifying mutations on a global scale, and has spurred a renewed enthusiasm for classical genetic screens in model organisms. -
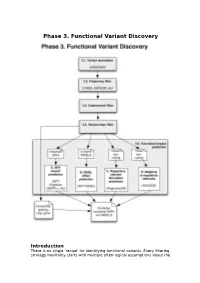
Phase 3. Functional Variant Discovery
Phase 3. Functional Variant Discovery Introduction There is no single ‘recipe’ for identifying functional variants. Every filtering strategy inevitably starts with multiple often-logical assumptions about the properties of causal variants that may also discard candidates too early. Novelty is one example, where the variant is not expected to be present in public repositories, even though apparently healthy individuals carrying it may have been sequenced in unrelated studies. High predicted functional impact is another, where a variant is expected to simultaneously occur at an evolutionarily conserved genomic location and also be scored as deleterious by multiple predictive algorithms may even be too stringent a rule in a Mendelian trait. In a similar vein, in a population genetics study, a novel amino acid class-changing variant that is not predicted to have a functional effect by the standard tools may have a molecular effect. An inheritance model may itself be incorrect, e.g. a condition may be autosomal recessive rather than X-linked as hypothesized, or digenic rather than recessive, etc. It is therefore important not to conclude too early that the variant(s) of interest “are not in the exome” and to exhaust all alternate plausible rules and scenarios before that conclusion is reached. Using disease variant discovery as a context, we aim to present here a range of guidelines that can be used to develop an appropriate variant prioritisation strategy. This phase will be presented as 5 sub-phases: 1. Variant annotation - using ANNOVAR (Wang et al, 2010) as an example 2. Frequency 3. Case-control filtering 4. Variant class filtering 5.