ATP-Binding Cassette Transporters G1 and G4 Mediate Cellular Cholesterol Efflux to High-Density Lipoproteins
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Pharmacogenomics of Vincristine-Induced Neurotoxicity
THE PHARMACOGENOMICS OF VINCRISTINE-INDUCED NEUROTOXICITY IN PAEDIATRIC CANCER PATIENTS WITH WILMS TUMOR OR RHABDOMYOSARCOMA by Tenneille Loo A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF SCIENCE in THE FACULTY OF GRADUATE STUDIES (Experimental Medicine) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) July 2011 © Tenneille Loo, 2011 Abstract Vincristine is one of the most effective and widely utilized antineoplastic agents. However, the clinical utility of this drug is limited by severely debilitating vincristine- induced neurotoxicities (VIN). Previous studies have associated VIN with genetic polymorphisms in genes involved in the metabolism and transportation of vincristine, including CYP3A4, CYP3A5, and ABCB1. However, the findings of such studies have not been consistently reproduced. This study hypothesizes that there are specific variants in genes involved in general drug absorption, metabolism, distribution, excretion, and toxicity (ADME-Tox) that affect the individual susceptibility to VIN in patients with Wilms tumor and rhabdomyosarcoma. Detailed clinical data was collected from 140 patients with Wilms tumor and rhabdomyosarcoma by retrospective chart review. VIN cases were characterized by type of neurotoxicity, and severity was evaluated using a validated clinical grading system for adverse events (NCI-CTCAE v4.03). A customized Illumina GoldenGate Panel was used to genotype 4,536 single nucleotide polymorphisms (SNPs) in candidate genes involved in the metabolism and transportation pathway of vincristine, as well as in genes broadly involved in ADME-Tox. None of the SNPs that were previously reported to be associated with VIN were found to be significantly associated (p-value < 0.05). With similar effect sizes, six novel genetic variants in five genes (PON1, ABCA4, ABCG1, CY51A1, SLCO1C1) were significantly associated with VIN in both tumor types. -

ABCG1 (ABC8), the Human Homolog of the Drosophila White Gene, Is a Regulator of Macrophage Cholesterol and Phospholipid Transport
ABCG1 (ABC8), the human homolog of the Drosophila white gene, is a regulator of macrophage cholesterol and phospholipid transport Jochen Klucken*, Christa Bu¨ chler*, Evelyn Orso´ *, Wolfgang E. Kaminski*, Mustafa Porsch-Ozcu¨ ¨ ru¨ mez*, Gerhard Liebisch*, Michael Kapinsky*, Wendy Diederich*, Wolfgang Drobnik*, Michael Dean†, Rando Allikmets‡, and Gerd Schmitz*§ *Institute for Clinical Chemistry and Laboratory Medicine, University of Regensburg, 93042 Regensburg, Germany; †National Cancer Institute, Laboratory of Genomic Diversity, Frederick, MD 21702-1201; and ‡Departments of Ophthalmology and Pathology, Columbia University, Eye Research Addition, New York, NY 10032 Edited by Jan L. Breslow, The Rockefeller University, New York, NY, and approved November 3, 1999 (received for review June 14, 1999) Excessive uptake of atherogenic lipoproteins such as modified low- lesterol transport. Although several effector molecules have been density lipoprotein complexes by vascular macrophages leads to proposed to participate in macrophage cholesterol efflux (6, 9), foam cell formation, a critical step in atherogenesis. Cholesterol efflux including endogenous apolipoprotein E (10) and the cholesteryl mediated by high-density lipoproteins (HDL) constitutes a protective ester transfer protein (11), the detailed molecular mechanisms mechanism against macrophage lipid overloading. The molecular underlying cholesterol export in these cells have not yet been mechanisms underlying this reverse cholesterol transport process are characterized. currently not fully understood. To identify effector proteins that are Recently, mutations of the ATP-binding cassette (ABC) trans- involved in macrophage lipid uptake and release, we searched for porter ABCA1 gene have been causatively linked to familial HDL genes that are regulated during lipid influx and efflux in human deficiency and Tangier disease (12–14). -

Genome-Wide Analysis of ATP-Binding
Tian et al. BMC Genomics (2017) 18:330 DOI 10.1186/s12864-017-3706-6 RESEARCH ARTICLE Open Access Genome-wide analysis of ATP-binding cassette (ABC) transporters in the sweetpotato whitefly, Bemisia tabaci Lixia Tian1, Tianxue Song2, Rongjun He1, Yang Zeng1, Wen Xie1, Qingjun Wu1, Shaoli Wang1, Xuguo Zhou3* and Youjun Zhang1* Abstract Background: ABC transporter superfamily is one of the largest and ubiquitous groups of proteins. Because of their role in detoxification, insect ABC transporters have gained more attention in recent years. In this study, we annotated ABC transporters from a newly sequenced sweetpotato whitefly genome. Bemisia tabaci Q biotype is an emerging global invasive species that has caused extensive damages to field crops as well as ornamental plants. Results: A total of 55 ABC transporters containing all eight described subfamilies (A to H) were identified in the B. tabaci Q genome, including 8 ABCAs, 3 ABCBs, 6 ABCCs, 2 ABCDs, 1 ABCE, 3 ABCFs, 23 ABCGs and 9 ABCHs. In comparison to other species, subfamilies G and H in both phloem- and blood-sucking arthropods are expanded. The temporal expression profiles of these 55 ABC transporters throughout B. tabaci developmental stages and their responses to imidacloprid, a neonicotinoid insecticide, were investigated using RNA-seq analysis. Furthermore, the mRNA expression of 24 ABC transporters (44% of the total) representing all eight subfamilies was confirmed by the quantitative real-time PCR (RT-qPCR). Furthermore, mRNA expression levels estimated by RT-qPCR and RNA-seq analyses were significantly correlated (r =0.684,p <0.01). Conclusions: It is the first genome-wide analysis of the entire repertoire of ABC transporters in B. -

Evolutionary and Functional Studies on Microsporidian ATP-Binding
Infection, Genetics and Evolution 68 (2019) 136–144 Contents lists available at ScienceDirect Infection, Genetics and Evolution journal homepage: www.elsevier.com/locate/meegid Evolutionary and functional studies on microsporidian ATP-binding T cassettes: Insights into the adaptation of microsporidia to obligated intracellular parasitism Qiang Hea, Charles R. Vossbrinckc, Qiong Yangd, Xian-Zhi Menga, Jian Luoa, Guo-Qing Pana, ⁎ ⁎ Ze-Yang Zhoua,b, , Tian Lia, a State Key Laboratory of Silkworm Genome Biology, Southwest University, Chongqing 400715, China b College of Life Science, Chongqing Normal University, Chongqing 400047, China c Department of Soil and Water, The Connecticut Agricultural Experiment Station, 123 Huntington Street, New Haven, CT 06511, USA d Sericulture and Agri-Food Research Institute, Guangdong Academy of Agricultural Sciences, Guangzhou, China ARTICLE INFO ABSTRACT Keywords: ATP-binding cassette (ABC) transporters comprise the largest family of transmembrane proteins and are found in Microsporidian all domains of life. The ABCs are involved in a variety of biological processes and as exporters play important ABC transporter roles in multidrug resistance. However, the ABC transporters have not been addressed in microsporidia, which Genome are a very large group of obligate intracellular parasites that can infect nearly all animals, including humans. Evolution Here, a total of 234 ABC transporters were identified from 18 microsporidian genomes and classified intofive Nosema bombycis subfamilies, including 74 ABCBs, 2 ABCCs, 18 ABCEs, 15 ABCFs, 102 ABCGs and 23 uncategorized members. Two subfamilies, ABCA and ABCD, are found in most organisms, but lost in microsporidia. Phylogenetic analysis indicated that microsporidian ABCB and ABCG subfamilies expanded by recent gene duplications, which re- sulted in the two largest subfamilies in microsporidia. -

Polymorphisms of the Multidrug Pump ABCG2: a Systematic Review of Their Effect on Protein Expression, Function, and Drug Pharmacokinetics
1521-009X/46/12/1886–1899$35.00 https://doi.org/10.1124/dmd.118.083030 DRUG METABOLISM AND DISPOSITION Drug Metab Dispos 46:1886–1899, December 2018 Copyright ª 2018 by The American Society for Pharmacology and Experimental Therapeutics Minireview Polymorphisms of the Multidrug Pump ABCG2: A Systematic Review of Their Effect on Protein Expression, Function, and Drug Pharmacokinetics Niall Heyes, Parth Kapoor, and Ian D. Kerr School of Life Sciences, Queen’s Medical Centre, University of Nottingham, Nottingham, United Kingdom Received June 12, 2018; accepted September 20, 2018 Downloaded from ABSTRACT The widespread expression and polyspecificity of the multidrug to improve outcomes in cancer patients treated with tyrosine ABCG2 efflux transporter make it an important determinant of the kinase inhibitors, but the reasons for this are yet to be estab- pharmacokinetics of a variety of substrate drugs. Null ABCG2 lished, and this residue’s role in the mechanism of the protein is expression has been linked to the Junior blood group. Polymor- unexplored by current biochemical and structural approaches. phisms affecting the expression or function of ABCG2 may have Research into the less-common polymorphisms is confined to dmd.aspetjournals.org clinically important roles in drug disposition and efficacy. The in vitro studies, with several polymorphisms shown to decrease most well-studied single nucleotide polymorphism (SNP), Q141K resistance to anticancer agents such as SN-38 and mitoxantrone. (421C>A), is shown to decrease ABCG2 expression and activity, In this review, we present a systematic analysis of the effects of resulting in increased total drug exposure and decreased re- ABCG2 polymorphisms on ABCG2 function and drug pharmaco- sistance to various substrates. -

Identification of Candidate ATP-Binding Cassette Transporter
www.nature.com/scientificreports OPEN Identifcation of candidate ATP- binding cassette transporter gene family members in Diaphorina citri (Hemiptera: Psyllidae) via adult tissues transcriptome analysis Zhengbing Wang 1, Fajun Tian1, Lijun Cai2, Jie Zhang2, Jiali Liu1 & Xinnian Zeng1* The ATP-binding cassette (ABC) transporters exist in all living organisms and play major roles in various biological functions by transporting a wide variety of substrates across membranes. The functions of ABC transporters in drug resistance have been extensively studied in vertebrates; however, they are rarely characterized in agricultural pests. The Asian citrus psyllid, Diaphorina citri, is one of the most damaging pests of the Citrus genus because of its transmission of Huanglongbing, also known as Yellow Dragon disease. In this study, the next-generation sequencing technique was applied to research the ABC transporters of D. citri. Fifty-three ABC transporter genes were found in the RNA-Seq data, and among these ABC transporters, 4, 4, 5, 2, 1, 4, 18 and 15 ABC proteins belonged to the ABCA-ABCH subfamilies, respectively. Diferent expression profles of 52 genes between imidacloprid-resistant and imidacloprid-susceptible strains were studied by qRT-PCR; 5 ABCGs and 4 ABCHs were signifcantly upregulated in the imidacloprid-resistant strain. In addition, fve of the nine upregulated genes were widely expressed in adult tissues in spatial expression analysis. The results suggest that these genes may play key roles in this phenotype. In general, this study contributed to our current understanding of D. citri resistance to insecticides. Te ATP-binding cassette (ABC) transporter family is one of the largest families of membrane proteins and uni- versally exists in all living organisms on Earth1. -

Ligand, Receptor, and Cell Type–Dependent Regulation of ABCA1 and ABCG1 Mrna in Prostate Cancer Epithelial Cells
Published OnlineFirst June 16, 2009; DOI: 10.1158/1535-7163.MCT-09-0020 Published Online First on June 16, 2009 as 10.1158/1535-7163.MCT-09-0020 OF1 Ligand, receptor, and cell type–dependent regulation of ABCA1 and ABCG1 mRNA in prostate cancer epithelial cells Steven E. Trasino,1 Young S. Kim,2 that ABCA1 gene expression is differentially regulated by and Thomas T.Y. Wang1 synthetic and natural LXR ligands, possibly involving kinase mediated signal transduction. [Mol Cancer Ther 2009;8(7): 1 Diet, Genomics, and Immunology Laboratory, Beltsville Human OF1–12] Nutrition Research Center, Agricultural Research Service, U.S. Department of Agriculture, Beltsville, Maryland and 2Nutritional Sciences Research Group, Division of Cancer Introduction Prevention, National Cancer Institute, NIH, Bethesda, Maryland Advanced and metastatic prostate cancer (PCa) remains the most prominent cancer type and the second leading cause of Abstract all cancer deaths among males in the United States (1). Recent evidence suggests that the liver X receptor (LXR) Treatment of recurrent or advanced PCa with hormone ab- is a potential anticancer target in prostate carcinoma. lation therapy typically results in disease regression with There is little characterization, however, of which of the eventual recurrence of PCa with a more aggressive and two LXR isoforms, LXRα or LXRβ, regulates the LXR- untreatable phenotype (2). In the absence of any effective responsive genes ATP-binding cassette subfamily members long-term therapy and with such an overwhelming social A1 (ABCA1)andG1(ABCG1) in transformed prostatic ep- burden, characterization of novel targets for effective che- ithelial cells. In this study, small interfering RNA (siRNA) moprevention or treatment of PCa has been a priority for α β cancer researchers. -

ABCG4: a Novel Human White Family ABC-Transporter Expressed in the Brain and Eye
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Biochimica et Biophysica Acta 1591 (2002) 175–179 www.bba-direct.com Short sequence-paper ABCG4: a novel human white family ABC-transporter expressed in the brain and eye Susan Oldfield *, Christopher A. Lowry, Jon Ruddick, Stafford L. Lightman University Research Centre for Neuroendocrinology, BRI, University of Bristol, Marlborough Street, Bristol BS2 8HW, UK Received 24 July 2001; received in revised form 11 April 2002; accepted 24 May 2002 Abstract White family transporters are a group of ATP-binding cassette (ABC) proteins that show sequence homology to the Drosophila white gene product. The Drosophila protein is a subunit of heterodimeric transporters of precursors for eye-pigment synthesis. A novel, human member of this family (ABCG4) has been identified. Northern blotting shows that ABCG4 is expressed specifically in the brain and the eye. D 2002 Elsevier Science B.V. All rights reserved. Keywords: White gene; ABC-transporter; ABCG1; Sterol ATP-binding cassette (ABC) transporters form a large and its murine counterpart is known as ABCG1 or ABC8. superfamily of proteins that mediate the transport of a large Northern blotting has shown that the mRNAs encoding variety of diverse molecules across biological membranes in ABCG1 are present in several tissues, predominantly brain, an ATP-dependent manner [1]. The functional transporters spleen and lung [10]. contain two ATP-binding folds and two transmembrane The function of ABCG1 is unknown, although there is domains each with, usually, five or six membrane spanning evidence that suggests that it may have a role in cholesterol helices. -

Transcriptional and Post-Transcriptional Regulation of ATP-Binding Cassette Transporter Expression
Transcriptional and Post-transcriptional Regulation of ATP-binding Cassette Transporter Expression by Aparna Chhibber DISSERTATION Submitted in partial satisfaction of the requirements for the degree of DOCTOR OF PHILOSOPHY in Pharmaceutical Sciences and Pbarmacogenomies in the Copyright 2014 by Aparna Chhibber ii Acknowledgements First and foremost, I would like to thank my advisor, Dr. Deanna Kroetz. More than just a research advisor, Deanna has clearly made it a priority to guide her students to become better scientists, and I am grateful for the countless hours she has spent editing papers, developing presentations, discussing research, and so much more. I would not have made it this far without her support and guidance. My thesis committee has provided valuable advice through the years. Dr. Nadav Ahituv in particular has been a source of support from my first year in the graduate program as my academic advisor, qualifying exam committee chair, and finally thesis committee member. Dr. Kathy Giacomini graciously stepped in as a member of my thesis committee in my 3rd year, and Dr. Steven Brenner provided valuable input as thesis committee member in my 2nd year. My labmates over the past five years have been incredible colleagues and friends. Dr. Svetlana Markova first welcomed me into the lab and taught me numerous laboratory techniques, and has always been willing to act as a sounding board. Michael Martin has been my partner-in-crime in the lab from the beginning, and has made my days in lab fly by. Dr. Yingmei Lui has made the lab run smoothly, and has always been willing to jump in to help me at a moment’s notice. -

Overall and Sex-Specific Associations Between Methylation of the ABCG1
Qin et al. Clinical Epigenetics (2019) 11:189 https://doi.org/10.1186/s13148-019-0784-0 RESEARCH Open Access Overall and sex-specific associations between methylation of the ABCG1 and APOE genes and ischemic stroke or other atherosclerosis-related traits in a sibling study of Chinese population Xueying Qin1*† , Jin Li1, Tao Wu1, Yiqun Wu1, Xun Tang1, Pei Gao1, Lin Li2, Mengying Wang1, Yao Wu1, Xiaowen Wang1, Dafang Chen1 and Yonghua Hu1*† Abstract Background: Identifying subjects with a high risk of ischemic stroke is fundamental for prevention of the disease. Both genetic and environmental risk factors contribute to ischemic stroke, but the underlying epigenetic mechanisms which mediate genetic and environmental risk effects are not fully understood. The aim of this study was to explore whether DNA methylation loci located in the ATP-binding cassette G1 (ABCG1) and apolipoprotein E (APOE)genes, both involved in the metabolism of lipids in the body, are related to ischemic stroke, using the Fangshan/Family-based Ischemic Stroke Study in China. We also tested if these CpG sites were associated with early signs of cardiovascular atherosclerosis (carotid intima–media thickness (cIMT), ankle–brachial index (ABI), and brachial–ankle pulse wave velocity (baPWV)). Results: DNA methylation at the cg02494239 locus in ABCG1 was correlated with ischemic stroke after adjusting for gender, previous history of diabetes and hypertension, smoking, drinking, body mass index, and blood lipid levels (above vs below mean, OR = 2.416, 95% CI 1.024–5.700, P =0.044;75–100% percentile vs 0–25% percentile, OR = 4.461, 95% CI 1.226–16.225, P = 0.023). -
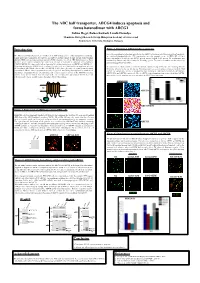
The ABC Half Transporter, ABCG4 Induces Apoptosis and Forms Heterodimer with ABCG1
The ABC half transporter, ABCG4 induces apoptosis and forms heterodimer with ABCG1 Zoltán Hegyi, Balázs Sarkadi, László Homolya Membrane Biology Research Group, Hungarian Academy of Sciences and Semmelweis University, Budapest, Hungary Introduction Figure 3. Functional ABCG4 induces apoptosis Since the morphological alterations observed in the ABCG4-expressing cells were indicative of apoptosis, The ABCG1 and ABCG4 proteins are members of the ATP binding cassette (ABC) transporter G subfamily. we examined phosphatidyl-serine (PS) translocation, an early apoptotic event in HEK293H cell lines Unlike most ABC transporters, the ABCG1 and ABCG4 proteins consist of only one nucleotide binding transiently transfected with the wt ABCG4 and its inactive mutant (KM) variant. PS translocation was domain (NBD) and one transmembrane domain (TMD), therefore are called ABC half-transporters. Some monitored by fluorescently labeled Annexin V binding (green). The total cell number was determined by members of the ABCG subfamily have proven to function as homodimers (ABCG2) or heterodimers nuclear staining by Hoechst (blue). (ABCG5/ABCG8). Previous results indicated potential heterodimerization between ABCG1 and ABCG4 (1). Regarding their function, ABCG1 has been proposed to play a role in cellular lipid/sterol regulation, whereas We found that the cultures transfected with wt ABCG4 contained a large number of cells exhibiting Annexin the function of ABCG4, the closest relative of ABCG1, is still elusive. Recently, we reported that functional V binding, whereas hardly any labeling for PS translocation was seen in cultures transfected with the KM expression of ABCG1 induces apoptosis in several cell types (2). Our finding was supported by rounded cell mutant. For comparison, Annexin V binding was also examined in cells transfected with the wt ABCG1, morphology, phosphatidyl-serine externalization, and elevated caspase 3 activity in the ABCG1-expressing ABCG1-KM, and ABCG2, respectively. -

Interindividual Differences in the Expression of ATP-Binding
Supplemental material to this article can be found at: http://dmd.aspetjournals.org/content/suppl/2018/02/02/dmd.117.079061.DC1 1521-009X/46/5/628–635$35.00 https://doi.org/10.1124/dmd.117.079061 DRUG METABOLISM AND DISPOSITION Drug Metab Dispos 46:628–635, May 2018 Copyright ª 2018 by The American Society for Pharmacology and Experimental Therapeutics Special Section on Transporters in Drug Disposition and Pharmacokinetic Prediction Interindividual Differences in the Expression of ATP-Binding Cassette and Solute Carrier Family Transporters in Human Skin: DNA Methylation Regulates Transcriptional Activity of the Human ABCC3 Gene s Tomoki Takechi, Takeshi Hirota, Tatsuya Sakai, Natsumi Maeda, Daisuke Kobayashi, and Ichiro Ieiri Downloaded from Department of Clinical Pharmacokinetics, Graduate School of Pharmaceutical Sciences, Kyushu University, Fukuoka, Japan (T.T., T.H., T.S., N.M., I.I.); Drug Development Research Laboratories, Kyoto R&D Center, Maruho Co., Ltd., Kyoto, Japan (T.T.); and Department of Clinical Pharmacy and Pharmaceutical Care, Graduate School of Pharmaceutical Sciences, Kyushu University, Fukuoka, Japan (D.K.) Received October 19, 2017; accepted January 30, 2018 dmd.aspetjournals.org ABSTRACT The identification of drug transporters expressed in human skin and levels. ABCC3 expression levels negatively correlated with the methylation interindividual differences in gene expression is important for understanding status of the CpG island (CGI) located approximately 10 kilobase pairs the role of drug transporters in human skin. In the present study, we upstream of ABCC3 (Rs: 20.323, P < 0.05). The reporter gene assay revealed evaluated the expression of ATP-binding cassette (ABC) and solute carrier a significant increase in transcriptional activity in the presence of CGI.