CPIMS 11 PI Research Meeting Condensed Phase and Interfacial
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Government Gazette Staatskoerant REPUBLIC of SOUTH AFRICA REPUBLIEK VAN SUID-AFRIKA LEGAL NOTICES WETLIKE KENNISGEWINGS A
Government Gazette Staatskoerant REPUBLIC OF SOUTH AFRICA REPUBLIEK VAN SUID-AFRIKA December Vol. 630 Pretoria, 1 2017 Desember No. 41282 PART 1 OF 2 LEGAL NOTICES A WETLIKE KENNISGEWINGS ISSN 1682-5843 N.B. The Government Printing Works will 41282 not be held responsible for the quality of “Hard Copies” or “Electronic Files” submitted for publication purposes 9 771682 584003 AIDS HELPLINE: 0800-0123-22 Prevention is the cure 2 No. 41282 GOVERNMENT GAZETTE, 1 DECEMBER 2017 IMPORTANT NOTICE: THE GOVERNMENT PRINTING WORKS WILL NOT BE HELD RESPONSIBLE FOR ANY ERRORS THAT MIGHT OCCUR DUE TO THE SUBMISSION OF INCOMPLETE / INCORRECT / ILLEGIBLE COPY. NO FUTURE QUERIES WILL BE HANDLED IN CONNECTION WITH THE ABOVE. Table of Contents LEGAL NOTICES BUSINESS NOTICES • BESIGHEIDSKENNISGEWINGS Gauteng ....................................................................................................................................... 12 Eastern Cape / Oos-Kaap ................................................................................................................. 13 Free State / Vrystaat ........................................................................................................................ 13 Western Cape / Wes-Kaap ................................................................................................................ 13 COMPANY NOTICES • MAATSKAPPYKENNISGEWINGS Gauteng ....................................................................................................................................... 14 LIQUIDATOR’S -
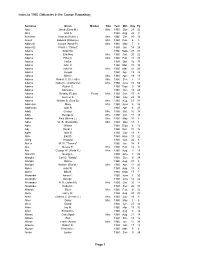
Index to 1960 Obituaries in the Canton Repository Page 1
Index to 1960 Obituaries in the Canton Repository Surname Given Maiden Title Year Mth Day Pg Abel Jacob (Clara M.) Mrs. 1960 Dec 28 32 Abel John A. 1960 Aug. 29 9 Ackelson Thomas (Ruth I.) Mrs. 1960 Dec 10 10 Ackert Edward (Patience) Mrs. 1960 Feb. 6 8 Adamcik Joseph (Anna P.) Mrs. 1960 May 7 6 Adametz Frank J. "Chant" 1960 Jan. 14 29 Adams Bright M. 1960 Sept. 28 28 Adams Ella May Mrs. 1960 Oct. 20 22 Adams Ethel A. Mrs. 1960 Feb. 15 20 Adams Helen 1960 Oct. 16 37 Adams John H. 1960 Mar. 31 39 Adams John W. Mrs. 1960 Mar. 21 26 Adams Joseph 1960 Apr. 13 24 Adams Minnie Mrs. 1960 Apr. 19 15 Adams Robert C. (Celestie) Mrs. 1960 Dec 3 11 Adams Robert L. (Catherine) Mrs. 1960 June 15 56 Adams Robert S. 1960 Sept. 9 14 Adams Samuel L. 1960 Jan. 19 20 Adams Schalto (Retta) Peary Mrs. 1960 Oct. 15 8 Adams Sumner S. 1960 July 28 10 Adams William B. (Zula B.) Mrs. 1960 Aug. 23 34 Adamson Mary Mrs. 1960 June 5 36 Addleman John R. 1960 Apr. 6 23 Addy George Mrs. 1960 Oct. 16 38 Addy George D. Mrs. 1960 Oct. 17 18 Adkins Paul (Wilma L.) Mrs. 1960 May 10 8 Adler WR(DllMW. R. (Della May ) Mrs. 1960 May 17 8 Adler William 1960 Sept. 6 16 Ady Oscar J. 1960 Dec. 31 12 Agler John B. 1960 July 19 8 Ahle Earl D. 1960 Nov. 18 22 Ailing Howard 1960 Oct. -
Bdo International Directory 2017
International Directory 2017 Latest version updated 5 July 2017 1 ABOUT BDO BDO is an international network of public accounting, tax and advisory firms, the BDO Member Firms, which perform professional services under the name of BDO. Each BDO Member Firm is a member of BDO International Limited, a UK company limited by guarantee. The BDO network is governed by the Council, the Global Board and the Executive (or Global Leadership Team) of BDO International Limited. Service provision within the BDO network is coordinated by Brussels Worldwide Services BVBA, a limited liability company incorporated in Belgium with VAT/BTW number BE 0820.820.829, RPR Brussels. BDO International Limited and Brussels Worldwide Services BVBA do not provide any professional services to clients. This is the sole preserve of the BDO Member Firms. Each of BDO International Limited, Brussels Worldwide Services BVBA and the member firms of the BDO network is a separate legal entity and has no liability for another such entity’s acts or omissions. Nothing in the arrangements or rules of BDO shall constitute or imply an agency relationship or a partnership between BDO International Limited, Brussels Worldwide Services BVBA and/or the member firms of the BDO network. BDO is the brand name for the BDO network and all BDO Member Firms. BDO is a registered trademark of Stichting BDO. © 2017 Brussels Worldwide Services BVBA 2 2016* World wide fee Income (millions) EUR 6,844 USD 7,601 Number of countries 158 Number of offices 1,401 Partners 5,736 Professional staff 52,486 Administrative staff 9,509 Total staff 67,731 Web site: www.bdointernational.com (provides links to BDO Member Firm web sites world wide) * Figures as per 30 September 2016 including exclusive alliances of BDO Member Firms. -

Coherence in Molecular Photoionization
Coherence in Molecular Photoionization PhD thesis David Ayuso Molinero Supervisors: ○ Alicia Palacios Cañas ○ Fernando Martín García arXiv:1705.06276v1 [physics.chem-ph] 17 May 2017 December 2015 Coherence in Molecular Photoionization PhD thesis David Ayuso Abstract We present a theoretical study of light-induced phenomena in gas-phase molecu- les, exploring the physical phenomena arising in two distinct contexts: using syn- chrotron radiation and ultrashort laser pulses. This work has been done in close collaboration with Piero Decleva (Universit`adegli Studi di Trieste). We first present our work on inner-shell photoionization of diatomic (CO) and small polyatomic (CF4, BF3) molecules at high photoelectron energies performed in collaboration with the experimental the groups of Edwin Kukk (Turku Univer- sity), Catalin Miron (Synchrotron SOLEIL), Kiyosi Ueda (Synchrotron SPring-8) and Thomas Darrah Thomas (Oregon State University). The combination of state-of-the-art Density Functional Theory (DFT)-like calculations, capable to describe photoionization accounting for the nuclear degrees of freedom, together with high-resolution third-generation synchrotron facilities, has enabled the inves- tigation of non-Franck-Condon effects observable in vibrationally resolved pho- toionization measurements. We demonstrate that the nuclear response to in- tramolecular electron diffraction is observable and can be used to obtain struc- tural information. As a proof-of-principle, by using the DFT calculations as an analysis tool to fit the experimental data, we have accurately determined the equi- librium distance of the CO molecule and the bond contraction that takes place upon C 1s ionization. This is a surplus of photoelectron spectroscopy with res- pect to more conventional spectroscopic techniques, which usually can only pro- vide structural information of neutral molecular species. -
Grace Phelan Final/Diss
UNIVERSITY OF CALIFORNIA SANTA CRUZ ANTOINE FRANÇOIS MOMORO “First Printer of National Liberty” 1756-1794 A dissertation submitted in partial satisfaction of the requirements for the degree of DOCTOR OF PHILOSOPHY in HISTORY by Grace M. Phelan September 2015 The Dissertation of Grace M. Phelan is approved: _______________________________ Professor Jonathan Beecher, Chair _______________________________ Professor Mark Traugott _______________________________ Professor Marilyn Westerkamp ___________________________________ Tyrus Miller Vice Provost and Dean of Graduate Studies Copyright © by Grace M. Phelan 2015 Table of Contents List of Illustrations iv Acknowledgments viii Introduction 1 1. From Besançon to Paris: Momoro's transition from Old Regime libraire to "First Printer of National Liberty" 14 2. Momoro's Political Ascendancy in Sectional Politics 84 3. Traité Elémentaire de l'Imprimerie: Conservative Instruction from the "First Printer of National Liberty" 177 4. Letters from the Vendée: Momoro's Narrative of Revolution and Counter- Revolution 252 Epilogue 321 Appendix A: Books Sold by Momoro, 1788-1790 325 Appendix B: Momoro's Publications, 1789-1793 338 Archival Sources 356 Works Cited 357 iii List of Illustrations 1. Premier Imprimeur de la Liberté Nationale 53 2. Second portrait of Momoro by Peronard 54 iv ABSTRACT Grace M. Phelan ANTOINE FRANCOIS MOMORO: "First Printer of National Liberty" 1756-1794 Antoine François Momoro (1756-1794) appears in historiographies of the French Revolution, in the history of printing and typography and in the history of work during the eighteenth century. Historians of the 1789 Revolution have often defined Momoro as either a sans-culottes or spokesman for the sans-culottes. Marxist historians and thinkers defined Momoro as an early socialist thinker for his controversial views on price fixing and private property. -
Traitement Position/Poste Taxable Benefits
Taxable Surname/Nom de Given Name/ Salary Paid/ Benefits/ Employer/Employeur famille Prénom Position/Poste Traitement Avant. impos. City of Oshawa SYMONS-MILROY CINDY Director, Economic Development Services$147,536.22 $660.50 City of Oshawa THURSTON GLENN Manager, Human Resource Services and Safety$108,684.19 $486.64 City of Oshawa THWAITES RODERICK Captain $108,428.43 $426.41 City of Oshawa TOOR MANMOHAN Manager, Construction Services $108,684.48 $486.64 City of Oshawa TUTTON CHRISTOPHER Captain $108,428.43 $426.41 City of Oshawa VINCENT GORDON Captain $102,781.12 $426.41 City of Oshawa WATSON GREGORY Captain $116,514.03 $426.41 City of Oshawa WEBSTER JOHN Firefighter $100,358.86 $370.62 City of Oshawa WHETHAM KENNETH Firefighter $104,162.21 $370.62 City of Oshawa WILSON NANCI Fire Prevention Officer $100,895.00 $426.41 City of Oshawa WILTON KEVIN Firefighter $106,196.34 $370.62 City of Oshawa WOOD GLEN Firefighter $116,762.41 $370.62 City of Oshawa WOOD SCOTT Training Officer $100,295.00 $426.41 City of Oshawa WYLIE J. NICOLE Director, Finance/Treasurer $133,328.26 $598.88 City of Ottawa ADAMS MICHAEL Garage Attendant-Starter Placer 1 $101,457.29 $1,294.89 City of Ottawa AGOCS IMRE Working Supervisor-Mechanic Inspector 1$100,308.87 $1,925.05 City of Ottawa AKESON JEFF Zone Supervisor $134,721.51 $916.32 City of Ottawa ALADAS MOTAZ Section Manager, Systems Communications & Video$117,918.87 $388.32 City of Ottawa ALBERT MARC Fire Prevention Officer $121,776.46 $1,470.50 City of Ottawa ALLAIRE LISA Chief, Corporate Communications $143,738.94 -

Surname Variations in Vermont Baptism Files by Town & Parish
These 5,704 Vermont Surname Variations are from the SEE's in our VT-FCGS Baptim Books. The Town-Church names show what area of Vermont they were used. Note: A few are the surname of a wife's previous marriage. Don't be fooled. Charlotte Mt Carmel Abair see Abare Vergennes St Peter Abair see Hebert Middlebury Assumption Abair see Hebert, Eubar Northfield St John Abaire see Hebert FairfieldBakersfield multi Abare see Hebert St Albans Holy Angels Abare see Hebert Waterbury St Andrew Abare see Hebert, Herbert Burlington Cath 1858-1930 Ablow see Ambleau Swanton-Highgate BVM-St Louis Adam see Laramee St Johnsbury Notre Dame Ahearn see Hearn Newport St Mary Aiken see Aitken, Arcand Burlington St Joseph Aikey see Ethier Burlington Cath 1858-1930 Aile see Ale Vergennes St Peter Ains see Hanks St Albans St Mary Ainse see Hanse, Hince, Hinse, Hance, Hains Charlotte Mt Carmel Ainse see Hens, Ens Middlebury Assumption Ainse see Hense, Hainse Burlington St Joseph Ainse see Hunt, Yantz Burlington St Joseph Ainsley see Thibault Bennington multi Akins see Daley Winooski St Stephen Aky see Etier Burlington Cath 1858-1930 Alam see Allen Burlington St Joseph Alapa see Arpin Burlington St Joseph Albarelli see Alberi Burlington Cath 1858-1930 Albarelli see Albary, Albery Burlington Cath 1858-1930 Albary see Albarelli, Albery Burlington St Joseph Alberi see Albarelli Rutland IHM Alberico see Albrigo Rutland IHM Albrigo see Alberico Vergennes St Peter Alden see Livernois Northfield St John Alden see Smith Lyndonville St Elizabeth Aldrich see Moore Burlington -

Lion Trophy Part 7
Conversation Contents FW: Zim Lion Hunting and Research Report - update 20160131 Attachments: /5. FW: Zim Lion Hunting and Research Report - update 20160131/1.1 ZIMBABWE LION HUNTING & RESEARCH REPORT USFWS BdP 20160131.pdf /5. FW: Zim Lion Hunting and Research Report - update 20160131/1.2 _Certification_.htm Pete Fick <[email protected]> From: Pete Fick <[email protected]> Sent: Thu Nov 23 2017 22:14:00 GMT-0700 (MST) To: <[email protected]>, <[email protected]> Subject: FW: Zim Lion Hunting and Research Report - update 20160131 ZIMBABWE LION HUNTING & RESEARCH REPORT USFWS Attachments: BdP 20160131.pdf _Certification_.htm Further info you may not have seen. From: Pete Fick [mailto:[email protected]] Sent: Friday, 11 March 2016 9:01 AM To: '[email protected]'; [email protected]; Meyers, Rachel L (Harare) ([email protected]) ([email protected]); [email protected]; [email protected]; [email protected]; [email protected]; Corkey, Christopher T (Harare) ([email protected]) ([email protected]); [email protected]; [email protected]; Plemons, Katherine L (Harare) ([email protected]) ([email protected]) Subject: FW: Zim Lion Hunting and Research Report - update 20160131 Hi All Please see attached lion report sent to USFWS (Tim Van Norman) in case you have not seen this. Regards Pete Conversation Contents USFWS Issues Permits For Lions From Zimbabwe and Zambia "Recce, Susan" <[email protected]> From: "Recce, Susan" <[email protected]> Sent: Mon Oct 23 2017 09:37:09 GMT-0600 (MDT) To: "'[email protected]'" <[email protected]> Subject: USFWS Issues Permits For Lions From Zimbabwe and Zambia Hi Greg, I wanted to see if the information below is correct and if there is anything available on this directly from the Service. -

Paris, 6-7 June
Paris, 6-7 June www.oecd.org/forum LastName FirstName JobTitle Company Country AALTO-MATTURI Sari Suomen Mielenterveysseura Executive director Finland AALTONEN Kristina DLI - Danish Teacher Trade Unions Head of office Belgium AARON Carl EWMI Direcctor United Kingdom AARUP Knud Knud Aarup Consulting Independent agent in developing welfare states Denmark ABBASOVA Adile WPL Azerbaijan ABBASZADEH Babak Toronto Centre President & CEO Canada ABBOTT Eva The British Embassy in Paris Communications Officer France Secrétariat Général des Affaires ABBOU Edmond Européennes (SGAE) Chef du secteur OCDE/SGAE France ABBOUD Leila Bloomberg News Tech Columnist France ABDALLAH Amr Education for Employment Foundaiton Senior Program Manager United States ABDELREHEM Muhammad Sciences Po / NODES Policy Incubator MPA Candidate /Chief Innovation Officer (CIO) France ABDELSADOK NEE BENCHABA Djemaia Assemblée populaire algérienne parlementaire Algeria ABDERRAZAK Mohamed Jouneidi Ministry of Industry, Energy and Mines Economist Tunisia ABDO Nouran Observatory of Higher Education Executive Manager Egypt Administration générale des impôts et des domaines (AGID) du Ministère des Finances ABDOURAHAMANE Mohamed Moneim des Comores Chef de service central informatique et statistique Comoros Central Research Institute of Electric Power ABE Kazuma Industry Nuclear Japan ABELA Carolyne AUSTRADE - Australian Trade Commission Investment Manager, Western Europe France ABISSEGUE Julie D'JU Agency - Afropean ChangeMaker Founder France ABIYEVA-VALMIER Karlygash Sciences Po Paris-CERI