Coherence in Molecular Photoionization
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Technical Application Note
Compact Rugged Spectrometers - A Universe of Spectroscopy Systems Technical Application Note Vibrational Spectroscopy: Infrared vs. Raman StellarNet, Inc. Tampa, Fl USA Tony Rizzuto, PhD April 12, 2017 Vibrational spectroscopies are integral in analyzing some of the most fundamentally important processes in physical chemistry: molecular vibrations. While there are many different experimental techniques used to analyze those vibrations, most are variations of the “Big Two,” FTIR and Raman scattering spectroscopies. This article aims to analyze the benefits and drawbacks of each of the “Big Two,” with the hope that it helps one choose the correct technique for one’s research interests. 1. What are the “Big Two?” Fourier Transform Infrared Spectroscopy Raman spectroscopy relies on inelastic (FTIR) is a simple absorption measurement scattering phenomenon that probes the where the detector measures the molecular vibration. Where FTIR will use a absorbance of infrared radiation by the broadband IR source, Raman spectroscopy sample. Each sample will absorb different typically uses a narrow-band, amounts of each frequency resulting in a monochromatic light source in order to “chemical fingerprint” that is the FTIR excite the vibrations of the molecule in your spectrum. sample. 2. Selection Rules: What molecular vibrations are being probed? Imagining a molecular bond vibration with the traditional ball and spring model (Figure 1a) and its resultant harmonic oscillator depiction (Figure 1b) allows us to calculate the selection rules for vibrational transitions to be = 1. However, that does not tell the whole story. In order to truly differentiate FTIR from Raman Spectroscopy, we must think about it on a molecular level. Compact Rugged Spectrometers - A Universe of Spectroscopy Systems Technical Application Note A B Figure 1: A. -

Government Gazette Staatskoerant REPUBLIC of SOUTH AFRICA REPUBLIEK VAN SUID-AFRIKA LEGAL NOTICES WETLIKE KENNISGEWINGS A
Government Gazette Staatskoerant REPUBLIC OF SOUTH AFRICA REPUBLIEK VAN SUID-AFRIKA December Vol. 630 Pretoria, 1 2017 Desember No. 41282 PART 1 OF 2 LEGAL NOTICES A WETLIKE KENNISGEWINGS ISSN 1682-5843 N.B. The Government Printing Works will 41282 not be held responsible for the quality of “Hard Copies” or “Electronic Files” submitted for publication purposes 9 771682 584003 AIDS HELPLINE: 0800-0123-22 Prevention is the cure 2 No. 41282 GOVERNMENT GAZETTE, 1 DECEMBER 2017 IMPORTANT NOTICE: THE GOVERNMENT PRINTING WORKS WILL NOT BE HELD RESPONSIBLE FOR ANY ERRORS THAT MIGHT OCCUR DUE TO THE SUBMISSION OF INCOMPLETE / INCORRECT / ILLEGIBLE COPY. NO FUTURE QUERIES WILL BE HANDLED IN CONNECTION WITH THE ABOVE. Table of Contents LEGAL NOTICES BUSINESS NOTICES • BESIGHEIDSKENNISGEWINGS Gauteng ....................................................................................................................................... 12 Eastern Cape / Oos-Kaap ................................................................................................................. 13 Free State / Vrystaat ........................................................................................................................ 13 Western Cape / Wes-Kaap ................................................................................................................ 13 COMPANY NOTICES • MAATSKAPPYKENNISGEWINGS Gauteng ....................................................................................................................................... 14 LIQUIDATOR’S -

Neutron Vibrational Spectroscopy
Neutron Vibrational Spectroscopy A.J. (Timmy) Ramirez-Cuesta Luke L. Daemen Yongqiang Cheng Spallation Neutron Source Oak Ridge National Laboratory ORNL is managed by UT-Battelle for the US Department of Energy The S(Q,w) Map w=0 Elastic Scattering Diffraction Structural Information 2 "Let there be light" 3 4 How to measure INS (1) Direct Geometry Instrumentation 4000 3500 Direct geometry instruments 3000 ) 1 - m measure Q trajectory is c ( 2500 r e f determined by the angle s n 2000 a r t and energy transfer. y g r 1500 e n Examples: ARCS, CNCS, E HYSPEC, SEQUIOA 1000 500 0 0 10 20 30 Momentum transfer (A-1) ) . U . A ( y t i s n e L t 2 n I Incident neutron beam is 0 10 20 30 40 50 60 70 Energy (meV) s monochromatic t n u o c determining the incident n o r t u Distance energy E1. e N That determines T1. We L1 12000 13000 14000 15000 16000 measure the ToF and we ToF (ms) can work out T2. Resolution is almost constant in units of Ei time 5 VISION: a high throughput spectrometer for neutron vibrational spectroscopy Yongqiang Cheng, Luke Daemen, A.J. (Timmy) Ramirez-Cuesta Spallation Neutron Source, Oak Ridge National Laboratory, Oak Ridge, TN 37931 USA VISION offers users a variety of sample environments How to measure INS (2) Polybenzene nanothreads synthesized at high pressure: single nanothreads VISION is best thought of as and experimental/computational capabilities: the neutron analogue of a Structural inference through modeling of vibrational spectra Indirect Geometry Instrumentation Raman spectrometer. -

Femtosecond Time-Resolved Observation of Butterfly Vibration In
www.nature.com/scientificreports OPEN Femtosecond time-resolved observation of butterfy vibration in electronically excited Received: 7 June 2017 Accepted: 11 October 2017 o-fuorophenol Published: xx xx xxxx Fengzi Ling1,2, Shuai Li1,2, Xinli Song1,2, Yanmei Wang1,2, Jinyou Long1,2 & Bing Zhang 1,2 The butterfy vibration during the hydrogen tunneling process in electronically excited o-fuorophenol has been visualized in real time by femtosecond time-resolved ion yield spectroscopy coupled with time-resolved photoelectron imaging technique. A coherent superposition of out-of-plane C–F butterfy motions is prepared in the frst excited electronic state (S1). As the C–F bond vibrates with respect to the aromatic ring, the nuclear geometry varies periodically, leading to the corresponding variation in the photoionization channel. By virtue of the more favorable ionization probability from the nonplanar minimum via resonance with the Rydberg states, the evolution of the vibrational wave packet is manifested as a superimposed beat in the parent-ion transient. Moreover, time-resolved photoelectron spectra ofer a direct mapping of the oscillating butterfy vibration between the planar geometry and nonplanar minimum. The beats for the photoelectron peaks originating from the planar geometry are out of phase with those from the nonplanar minimum. Our results provide a physically intuitive and complete picture of the oscillatory fow of energy responsible for the coherent vibrational motion on the excited state surface. Ultrafast molecular vibration is one of the fundamental motions that characterize chemical bonding and decide reaction dynamics at the molecular level1–3. Over the past few decades, frequency-resolved spectroscopies such as infrared and Raman spectra4 and resonant two-photon ionization (R2PI) spectroscopy5 have devoted to pro- viding detailed spectroscopic data about molecular vibration. -

All-Digital Histopathology by Infrared-Optical Hybrid Microscopy
All-digital histopathology by infrared-optical hybrid microscopy Martin Schnella, Shachi Mittala,b, Kianoush Falahkheirkhaha,c, Anirudh Mittala,b, Kevin Yeha,b, Seth Kenkela,d, Andre Kajdacsy-Ballae, P. Scott Carneyf, and Rohit Bhargavaa,b,c,d,g,h,i,1 aBeckman Institute for Advanced Science and Technology, University of Illinois at Urbana–Champaign, Urbana, IL 61801; bDepartment of Bioengineering, University of Illinois at Urbana–Champaign, Urbana, IL 61801; cDepartment of Chemical and Biomolecular Engineering, University of Illinois at Urbana– Champaign, Urbana, IL 61801; dDepartment of Mechanical Science and Engineering, University of Illinois at Urbana–Champaign, Urbana, IL 61801; eDepartment of Pathology, University of Illinois at Chicago, Chicago, IL 60612; fThe Institute of Optics, University of Rochester, Rochester, NY 14620; gCancer Center at Illinois, University of Illinois at Urbana–Champaign, Urbana, IL 61801; hDepartment of Electrical and Computer Engineering, University of Illinois at Urbana–Champaign, Urbana, IL 61801; and iDepartment of Chemistry, University of Illinois at Urbana–Champaign, Urbana, IL 61801 Edited by Christian Huck, University of Innsbruck, Innsbruck, Austria, and accepted by Editorial Board Member John A. Rogers December 20, 2019 (received for review July 19, 2019) Optical microscopy for biomedical samples requires expertise in fidelity imaging in both point-scanning (15) and wide-field (16–19) staining to visualize structure and composition. Midinfrared (mid-IR) modalities, IR pixel sizes are ∼100-fold larger than those easily spectroscopic imaging offers label-free molecular recording and achieved in visible microscopy, and data acquisition speed is still virtual staining by probing fundamental vibrational modes of three to four orders of magnitude slower than in visible imaging molecular components. -
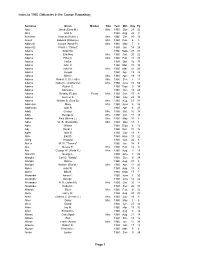
Index to 1960 Obituaries in the Canton Repository Page 1
Index to 1960 Obituaries in the Canton Repository Surname Given Maiden Title Year Mth Day Pg Abel Jacob (Clara M.) Mrs. 1960 Dec 28 32 Abel John A. 1960 Aug. 29 9 Ackelson Thomas (Ruth I.) Mrs. 1960 Dec 10 10 Ackert Edward (Patience) Mrs. 1960 Feb. 6 8 Adamcik Joseph (Anna P.) Mrs. 1960 May 7 6 Adametz Frank J. "Chant" 1960 Jan. 14 29 Adams Bright M. 1960 Sept. 28 28 Adams Ella May Mrs. 1960 Oct. 20 22 Adams Ethel A. Mrs. 1960 Feb. 15 20 Adams Helen 1960 Oct. 16 37 Adams John H. 1960 Mar. 31 39 Adams John W. Mrs. 1960 Mar. 21 26 Adams Joseph 1960 Apr. 13 24 Adams Minnie Mrs. 1960 Apr. 19 15 Adams Robert C. (Celestie) Mrs. 1960 Dec 3 11 Adams Robert L. (Catherine) Mrs. 1960 June 15 56 Adams Robert S. 1960 Sept. 9 14 Adams Samuel L. 1960 Jan. 19 20 Adams Schalto (Retta) Peary Mrs. 1960 Oct. 15 8 Adams Sumner S. 1960 July 28 10 Adams William B. (Zula B.) Mrs. 1960 Aug. 23 34 Adamson Mary Mrs. 1960 June 5 36 Addleman John R. 1960 Apr. 6 23 Addy George Mrs. 1960 Oct. 16 38 Addy George D. Mrs. 1960 Oct. 17 18 Adkins Paul (Wilma L.) Mrs. 1960 May 10 8 Adler WR(DllMW. R. (Della May ) Mrs. 1960 May 17 8 Adler William 1960 Sept. 6 16 Ady Oscar J. 1960 Dec. 31 12 Agler John B. 1960 July 19 8 Ahle Earl D. 1960 Nov. 18 22 Ailing Howard 1960 Oct. -

Infrared Spectroscopy: Theory
Online edition for students of organic chemistry lab courses at the University of Colorado, Boulder, Dept of Chem and Biochem. (2002) u Chapter 15 Infrared Spectroscopy: Theory An important tool of the organic chemist is Infrared Spectroscopy, or IR. IR spectra are acquired on a special instrument, called an IR spectrometer. IR is used both to gather information about the structure of a compound and as an analytical tool to assess the purity of a compound. IR spectra are quick and easy to run, and since the teaching labs are fortunate enough to have three of these instruments, students are encouraged to acquire spectra of compounds made in the laboratory throughout the first and second semesters of the lab course. 15.1 The Electromagnetic Spectrum Infrared refers to that part of the electromagnetic spectrum between the visible and microwave regions. Electromagnetic spectrum refers to the seemingly diverse col- lection of radiant energy, from cosmic rays to X-rays to visible light to microwaves, each of which can be considered as a wave or particle traveling at the speed of light. These waves differ from each other in the length and frequency, as illustrated in Figure 15.1. Frequency, ν (nu), is the number of wave cycles that pass through a point in one sec- ond. It is measured in Hz, where 1 Hz = 1 cycle/sec. Wavelength, λ (lambda), is the length of one complete wave cycle. It is often measured in cm (centimeters). Wave- length and frequency are inversely related: c c ν = --- and λ = --- Eq. 1 λ ν where c is the speed of light, 3 x 1010 cm/sec Energy is related to wavelength and frequency by the following for- mulas: Eq. -

Two-Dimensional Infrared Spectroscopy of Vibrational Polaritons
Two-dimensional infrared spectroscopy of vibrational polaritons Bo Xianga, Raphael F. Ribeirob, Adam D. Dunkelbergerc, Jiaxi Wangb, Yingmin Lia, Blake S. Simpkinsc, Jeffrey C. Owrutskyc, Joel Yuen-Zhoub, and Wei Xionga,b,1 aMaterials Science and Engineering Program, University of California, San Diego, La Jolla, CA 92093; bDepartment of Chemistry and Biochemistry, University of California, San Diego, La Jolla, CA 92093; and cChemistry Division, Naval Research Laboratory, Washington, DC 20375 Edited by Stephen R. Forrest, University of Michigan, Ann Arbor, MI, and approved March 28, 2018 (received for review December 19, 2017) We report experimental 2D infrared (2D IR) spectra of coherent molecular-cavity system has many dark states in comparison with light–matter excitations––molecular vibrational polaritons. The ap- the few bright modes (3, 17, 18): One type of dark states shares plication of advanced 2D IR spectroscopy to vibrational polaritons molecular components and some small photonic character with challenges and advances our understanding in both fields. First, polaritons as a result of disorder (17). Another type of dark the 2D IR spectra of polaritons differ drastically from free uncoupled states, termed “uncoupled” molecules, consists of molecular vi- excitations and a new interpretation is needed. Second, 2D IR brations which are not coupled to the cavity electric field (e.g., uniquely resolves excitation of hybrid light–matter polaritons and because they may be outside the photonic-mode volume, or unexpected dark states in a state-selective manner, revealing other- because their transition dipole moment is orthogonal to the wise hidden interactions between them. Moreover, 2D IR signals mode polarization). -
Bdo International Directory 2017
International Directory 2017 Latest version updated 5 July 2017 1 ABOUT BDO BDO is an international network of public accounting, tax and advisory firms, the BDO Member Firms, which perform professional services under the name of BDO. Each BDO Member Firm is a member of BDO International Limited, a UK company limited by guarantee. The BDO network is governed by the Council, the Global Board and the Executive (or Global Leadership Team) of BDO International Limited. Service provision within the BDO network is coordinated by Brussels Worldwide Services BVBA, a limited liability company incorporated in Belgium with VAT/BTW number BE 0820.820.829, RPR Brussels. BDO International Limited and Brussels Worldwide Services BVBA do not provide any professional services to clients. This is the sole preserve of the BDO Member Firms. Each of BDO International Limited, Brussels Worldwide Services BVBA and the member firms of the BDO network is a separate legal entity and has no liability for another such entity’s acts or omissions. Nothing in the arrangements or rules of BDO shall constitute or imply an agency relationship or a partnership between BDO International Limited, Brussels Worldwide Services BVBA and/or the member firms of the BDO network. BDO is the brand name for the BDO network and all BDO Member Firms. BDO is a registered trademark of Stichting BDO. © 2017 Brussels Worldwide Services BVBA 2 2016* World wide fee Income (millions) EUR 6,844 USD 7,601 Number of countries 158 Number of offices 1,401 Partners 5,736 Professional staff 52,486 Administrative staff 9,509 Total staff 67,731 Web site: www.bdointernational.com (provides links to BDO Member Firm web sites world wide) * Figures as per 30 September 2016 including exclusive alliances of BDO Member Firms. -

Infrared Spectroscopy: Fundamentals and Applications
Contents Series Preface ix Preface xi Acronyms, Abbreviations and Symbols xiii About the Author xvii 1 Introduction 1 1.1 Electromagnetic Radiation 2 1.2 Infrared Absorptions 5 1.3 Normal Modes of Vibration 6 1.4 Complicating Factors 11 1.4.1 Overtone and Combination Bands 11 1.4.2 Fermi Resonance 12 1.4.3 Coupling 12 1.4.4 Vibration–Rotation Bands 12 References 13 2 Experimental Methods 15 2.1 Introduction 15 2.2 Dispersive Infrared Spectrometers 16 2.3 Fourier-Transform Infrared Spectrometers 18 2.3.1 Michelson Interferometers 18 2.3.2 Sources and Detectors 19 2.3.3 Fourier-Transformation 20 2.3.4 Moving Mirrors 21 2.3.5 Signal-Averaging 22 vi Infrared Spectroscopy: Fundamentals and Applications 2.3.6 Advantages 23 2.3.7 Computers 23 2.3.8 Spectra 24 2.4 Transmission Methods 25 2.4.1 Liquids and Solutions 25 2.4.2 Solids 28 2.4.3 Gases 31 2.4.4 Pathlength Calibration 32 2.5 Reflectance Methods 33 2.5.1 Attenuated Total Reflectance Spectroscopy 33 2.5.2 Specular Reflectance Spectroscopy 35 2.5.3 Diffuse Reflectance Spectroscopy 36 2.5.4 Photoacoustic Spectroscopy 37 2.6 Microsampling Methods 38 2.7 Chromatography–Infrared Spectroscopy 41 2.8 Thermal Analysis–Infrared Spectroscopy 42 2.9 Other Techniques 43 References 44 3 Spectral Analysis 45 3.1 Introduction 45 3.2 Group Frequencies 46 3.2.1 Mid-Infrared Region 46 3.2.2 Near-Infrared Region 47 3.2.3 Far-Infrared Region 48 3.3 Identification 48 3.4 Hydrogen Bonding 49 3.5 Spectrum Manipulation 51 3.5.1 Baseline Correction 51 3.5.2 Smoothing 51 3.5.3 Difference Spectra 52 3.5.4 Derivatives -

Infrared Spectroscopy
23 chapter Infrared Spectroscopy Randy L. Wehling Department of Food Science and Technology, University of Nebraska, Lincoln, NE 68583-0919, USA [email protected] 23.1 Introduction 409 23.3.2 Applications of Mid-IR 23.2 Principles of IR Spectroscopy 409 Spectroscopy 411 23.2.1 The IR Region of the Electromagnetic 23.3.2.1 Absorption Bands of Organic Spectrum 409 Functional Groups 411 23.2.2 Molecular Vibrations 409 23.3.2.2 Presentation of Mid-IR 23.2.3 Factors Affecting the Frequency Spectra 411 of Vibration 409 23.3.2.3 Qualitative Applications 412 23.3 Mid-IR Spectroscopy 410 23.3.2.4 Quantitative Applications 412 23.3.1 Instrumentation 410 23.3.3 Raman Spectroscopy 412 23.3.1.1 Dispersive Instruments 410 23.4 Near-Infrared Spectroscopy 413 23.3.1.2 Fourier Transform 23.4.1 Principles 413 Instruments 410 23.4.1.1 Principles of Diffuse Reflection 23.3.1.3 Sample Handling Measurements 413 Techniques 411 S.S. Nielsen, Food Analysis, Food Science Texts Series, DOI 10.1007/978-1-4419-1478-1_23, 407 °c Springer Science+Business Media, LLC 2010 408 Part IV • Spectroscopy 23.4.1.2 Absorption Bands in the NIR 23.4.4 Qualitative Analysis by NIR Region 414 Spectroscopy 417 23.4.2 Instrumentation 415 23.4.5 Applications of NIR Spectroscopy 23.4.3 Quantitative Methods Using NIR to Food Analysis 417 Spectroscopy 416 23.5 Summary 418 23.4.3.1 Calibration Methods 23.6 Study Questions 418 Using Multiple Linear 23.7 References 419 Regression 417 23.4.3.2 Calibration Development Using Full Spectrum Methods 417 Chapter 23 • Infrared Spectroscopy 409 23.1 INTRODUCTION Infrared (IR) spectroscopy refers to measurement of the absorption of different frequencies of IR radia- tion by foods or other solids, liquids, or gases. -

The Possible Role of Molecular Vibration in Intracellular Signalling
https://www.scientificarchives.com/journal/journal-of-cellular-signaling Journal of Cellular Signaling Review Article The Possible Role of Molecular Vibration in Intracellular Signalling Werner Jaross* Institute of Clinical Chemistry and Laboratory Medicine, University Hospital Carl Gustav Carus, Technische Universitaet Dresden, Fetscherstrasse 74, 01307, Dresden, Germany *Correspondence should be addressed to Werner Jaross; [email protected] Received date: July 03, 2020, Accepted date: October 27, 2020 Copyright: © 2020 Jaross W. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract The exchange of information within the cell is extremely complex. Besides the well-studied chemical signalling, physical signalling is required to fulfil spatial and temporal aspects. The Golgi apparatus and the microtubule skeleton system are the decisive structures for numerous intracellular transport tasks . Close communication between the Golgi apparatus and the cell periphery is an absolute prerequisite for the well-directed positioning of structural elements. The majority of the substances that influence the cell from the membrane transmit the information to the intracellular destination via signal transduction pathways. It is discussed in detail that the transmission of information in both systems is based on emission and resonance of electromagnetic patterns in the infrared frequency range generated by the vibrations of the respective molecules. This radiation with fingerprint patterns must be coherent to activate enzymes. Coherence could be achieved by the chemical reactions of the molecules to be replaced or in the signal transduction pathways, by the phosphorylation of the transduction proteins.