Histamine H2- and H3-Receptor Antagonists: Synthesis and Characterization of Radio- Labelledand Fluorescent Pharmacological Tool
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
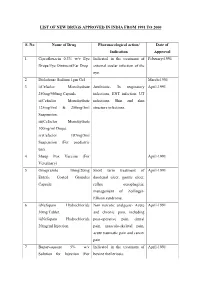
List of New Drugs Approved in India from 1991 to 2000
LIST OF NEW DRUGS APPROVED IN INDIA FROM 1991 TO 2000 S. No Name of Drug Pharmacological action/ Date of Indication Approval 1 Ciprofloxacin 0.3% w/v Eye Indicated in the treatment of February-1991 Drops/Eye Ointment/Ear Drop external ocular infection of the eye. 2 Diclofenac Sodium 1gm Gel March-1991 3 i)Cefaclor Monohydrate Antibiotic- In respiratory April-1991 250mg/500mg Capsule. infections, ENT infection, UT ii)Cefaclor Monohydrate infections, Skin and skin 125mg/5ml & 250mg/5ml structure infections. Suspension. iii)Cefaclor Monohydrate 100mg/ml Drops. iv)Cefaclor 187mg/5ml Suspension (For paediatric use). 4 Sheep Pox Vaccine (For April-1991 Veterinary) 5 Omeprazole 10mg/20mg Short term treatment of April-1991 Enteric Coated Granules duodenal ulcer, gastric ulcer, Capsule reflux oesophagitis, management of Zollinger- Ellison syndrome. 6 i)Nefopam Hydrochloride Non narcotic analgesic- Acute April-1991 30mg Tablet. and chronic pain, including ii)Nefopam Hydrochloride post-operative pain, dental 20mg/ml Injection. pain, musculo-skeletal pain, acute traumatic pain and cancer pain. 7 Buparvaquone 5% w/v Indicated in the treatment of April-1991 Solution for Injection (For bovine theileriosis. Veterinary) 8 i)Kitotifen Fumerate 1mg Anti asthmatic drug- Indicated May-1991 Tablet in prophylactic treatment of ii)Kitotifen Fumerate Syrup bronchial asthma, symptomatic iii)Ketotifen Fumerate Nasal improvement of allergic Drops conditions including rhinitis and conjunctivitis. 9 i)Pefloxacin Mesylate Antibacterial- In the treatment May-1991 Dihydrate 400mg Film Coated of severe infection in adults Tablet caused by sensitive ii)Pefloxacin Mesylate microorganism (gram -ve Dihydrate 400mg/5ml Injection pathogens and staphylococci). iii)Pefloxacin Mesylate Dihydrate 400mg I.V Bottles of 100ml/200ml 10 Ofloxacin 100mg/50ml & Indicated in RTI, UTI, May-1991 200mg/100ml vial Infusion gynaecological infection, skin/soft lesion infection. -

TABLE 1 Studies of Antagonist Activity in Constitutively Active
TABLE 1 Studies of antagonist activity in constitutively active receptors systems shown to demonstrate inverse agonism for at least one ligand Targets are natural Gs and constitutively active mutants (CAM) of GPCRs. Of 380 antagonists, 85% of the ligands demonstrate inverse agonism. Receptor Neutral Antagonist Inverse Agonist Reference Human β2-adrenergic Dichloroisoproterenol, pindolol, labetolol, timolol, Chidiac et al., 1996; Azzi et alprenolol, propranolol, ICI 118,551, cyanopindolol al., 2001 Turkey erythrocyte β-adrenergic Propranolol, pindolol Gotze et al., 1994 Human β2-adrenergic (CAM) Propranolol Betaxolol, ICI 118,551, sotalol, timolol Samama et al., 1994; Stevens and Milligan, 1998 Human/guinea pig β1-adrenergic Atenolol, propranolol Mewes et al., 1993 Human β1-adrenergic Carvedilol CGP20712A, metoprolol, bisoprolol Engelhardt et al., 2001 Rat α2D-adrenergic Rauwolscine, yohimbine, WB 4101, idazoxan, Tian et al., 1994 phentolamine, Human α2A-adrenergic Napthazoline, Rauwolscine, idazoxan, altipamezole, levomedetomidine, Jansson et al., 1998; Pauwels MPV-2088 (–)RX811059, RX 831003 et al., 2002 Human α2C-adrenergic RX821002, yohimbine Cayla et al., 1999 Human α2D-adrenergic Prazosin McCune et al., 2000 Rat α2-adrenoceptor MK912 RX821002 Murrin et al., 2000 Porcine α2A adrenoceptor (CAM- Idazoxan Rauwolscine, yohimbine, RX821002, MK912, Wade et al., 2001 T373K) phentolamine Human α2A-adrenoceptor (CAM) Dexefaroxan, (+)RX811059, (–)RX811059, RS15385, yohimbine, Pauwels et al., 2000 atipamezole fluparoxan, WB 4101 Hamster α1B-adrenergic -

Human Histamine H2 Receptor, Frozen Cells Product No.: ES-391-AF
TECHNICAL AequoZen® DATA SHEET Research use only. Not for use in diagnostic procedures. You are authorized to utilize these frozen cell preparations one time only. Any attempt to transfer, re-use, or propagate these cells is expressly unauthorized and a violation of the product terms and conditions of sale. Human Histamine H2 Receptor, Frozen Cells Product No.: ES-391-AF Lot No.: 2562845 Material Provided Cells: 1 x 1 mL frozen aliquot Format: ~10 x 106 cells/mL in Ham’s F12, 10% FBS with 10 % DMSO Product Information Cellular Background: CHO-K1 Parental Frozen Cells (control): A19 (replaced with Cat # ES-000-A2F) Frozen Cells Info: Frozen recombinant, CHO-K1 cells expressing mitochondrially- targeted Aequorin, Gα16 and the human Histamine H2 receptor. DNA Sequence: Identical to coding sequence of GenBank NM_022304.2. Corresponding Protein Sequence: Identical to GenBank NP_071640.1. Storage Conditions: Store in liquid nitrogen (vapor phase) immediately upon receipt, or maximum 15 days at -80°C. AequoZen® is designed for single use only. Do not refreeze. Quality Control ® EC50 for a reference agonist is determined using an AequoScreen assay (Figure 1). Mycoplasma test is performed using MycoAlert® Mycoplasma detection kit. We certify that these results meet our quality release criteria. Amthamine dihydrobromide (EC50): 6.9 nM Mycoplasma: This cell line tested negative for Mycoplasma. TDS-ES-391-AF-04 Page 1 of 5 Recommended Thawing Conditions and Handling of Frozen Cells Carefully follow instructions below to obtain the expected results. Most Frozen cells are intended to be assayed immediately upon thawing. Exceptionally, where specified, some frozen cell products require an overnight incubation in Cell Medium to enable them to perform optimally. -

The Histamine H4 Receptor: a Novel Target for Safe Anti-Inflammatory
GASTRO ISSN 2377-8369 Open Journal http://dx.doi.org/10.17140/GOJ-1-103 Review The Histamine H4 Receptor: A Novel Target *Corresponding author Maristella Adami, PhD for Safe Anti-inflammatory Drugs? Department of Neuroscience University of Parma Via Volturno 39 43125 Parma Italy * 1 Tel. +39 0521 903943 Maristella Adami and Gabriella Coruzzi Fax: +39 0521 903852 E-mail: [email protected] Department of Neuroscience, University of Parma, Via Volturno 39, 43125 Parma, Italy Volume 1 : Issue 1 1retired Article Ref. #: 1000GOJ1103 Article History Received: May 30th, 2014 ABSTRACT Accepted: June 12th, 2014 th Published: July 16 , 2014 The functional role of histamine H4 receptors (H4Rs) in the Gastrointestinal (GI) tract is reviewed, with particular reference to their involvement in the regulation of gastric mucosal defense and inflammation. 4H Rs have been detected in different cell types of the gut, including Citation immune cells, paracrine cells, endocrine cells and neurons, from different animal species and Adami M, Coruzzi G. The Histamine H4 Receptor: a novel target for safe anti- humans; moreover, H4R expression was reported to be altered in some pathological conditions, inflammatory drugs?. Gastro Open J. such as colitis and cancer. Functional studies have demonstrated protective effects of H4R an- 2014; 1(1): 7-12. doi: 10.17140/GOJ- tagonists in several experimental models of gastric mucosal damage and intestinal inflamma- 1-103 tion, suggesting a potential therapeutic role of drugs targeting this new receptor subtype in GI disorders, such as allergic enteropathy, Inflammatory Bowel Disease (IBD), Irritable Bowel Syndrome (IBS) and cancer. KEYWORDS: Histamine H4 receptor; Stomach; Intestine. -

From Inverse Agonism to 'Paradoxical Pharmacology' Richard A
International Congress Series 1249 (2003) 27-37 From inverse agonism to 'Paradoxical Pharmacology' Richard A. Bond*, Kenda L.J. Evans, Zsirzsanna Callaerts-Vegh Department of Pharmacological and Pharmaceutical Sciences, University of Houston, 521 Science and Research Bldg 2, 4800 Caltioun, Houston, TX 77204-5037, USA Received 16 April 2003; accepted 16 April 2003 Abstract The constitutive or spontaneous activity of G protein-coupled receptors (GPCRs) and compounds acting as inverse agonists is a recent but well-established phenomenon. Dozens of receptor subtypes for numerous neurotransmitters and hormones have been shown to posses this property. However, do to the apparently low percentage of receptors in the spontaneously active state, the physiologic relevance of these findings remains questionable. The possibility that the reciprocal nature of the effects of agonists and inverse agonists may extend to cellular signaling is discussed, and that this may account for the beneficial effects of certain p-adrenoceptor inverse agonists in the treatment of heart failure. © 2003 Elsevier Science B.V. All rights reserved. Keywords. Inverse agonism; GPCR; Paradoxical pharmacology 1. Brief history of inverse agonism at G protein-coupled receptors For approximately three-quarters of a century, ligands that interacted with G protein- coupled receptors (GPCRs) were classified either as agonists or antagonists. Receptors were thought to exist in a single quiescent state that could only induce cellular signaling upon agonist binding to the receptor to produce an activated state of the receptor. In this model, antagonists had no cellular signaling ability on their own, but did bind to the receptor and prevented agonists from being able to bind and activate the receptor. -

(12) Patent Application Publication (10) Pub. No.: US 2010/0221245 A1 Kunin (43) Pub
US 2010O221245A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2010/0221245 A1 Kunin (43) Pub. Date: Sep. 2, 2010 (54) TOPICAL SKIN CARE COMPOSITION Publication Classification (51) Int. Cl. (76) Inventor: Audrey Kunin, Mission Hills, KS A 6LX 39/395 (2006.01) (US) A6II 3L/235 (2006.01) A638/16 (2006.01) Correspondence Address: (52) U.S. Cl. ......................... 424/133.1: 514/533: 514/12 HUSCH BLACKWELL SANDERS LLP (57) ABSTRACT 4801 Main Street, Suite 1000 - KANSAS CITY, MO 64112 (US) The present invention is directed to a topical skin care com position. The composition has the unique ability to treat acne without drying out the user's skin. In particular, the compo (21) Appl. No.: 12/395,251 sition includes a base, an antibacterial agent, at least one anti-inflammatory agent, and at least one antioxidant. The (22) Filed: Feb. 27, 2009 antibacterial agent may be benzoyl peroxide. US 2010/0221 245 A1 Sep. 2, 2010 TOPCAL SKIN CARE COMPOSITION stay of acne treatment since the 1950s. Skin irritation is the most common side effect of benzoyl peroxide and other anti BACKGROUND OF THE INVENTION biotic usage. Some treatments can be severe and can leave the 0001. The present invention generally relates to composi user's skin excessively dry. Excessive use of some acne prod tions and methods for producing topical skin care. Acne Vul ucts may cause redness, dryness of the face, and can actually garis, or acne, is a common skin disease that is prevalent in lead to more acne. Therefore, it would be beneficial to provide teenagers and young adults. -

Pitolisant (Wakix) Reference Number: CP.PMN.221 Effective Date: 03.01.20 Last Review Date: 02.21 Line of Business: Commercial, HIM, Medicaid Revision Log
Clinical Policy: Pitolisant (Wakix) Reference Number: CP.PMN.221 Effective Date: 03.01.20 Last Review Date: 02.21 Line of Business: Commercial, HIM, Medicaid Revision Log See Important Reminder at the end of this policy for important regulatory and legal information. Description ® Wakix (pitolisant) is a selective histamine 3 (H3) receptor antagonist/inverse agonist. FDA Approved Indication(s) Wakix is indicated for the treatment of excessive daytime sleepiness (EDS) or cataplexy in adult patients with narcolepsy. Policy/Criteria Provider must submit documentation (such as office chart notes, lab results or other clinical information) supporting that member has met all approval criteria. It is the policy of health plans affiliated with Centene Corporation® that Wakix is medically necessary when the following criteria are met: I. Initial Approval Criteria A. Narcolepsy with Cataplexy (must meet all): 1. Diagnosis of narcolepsy with cataplexy; 2. Prescribed by or in consultation with a neurologist or sleep medicine specialist; 3. Age ≥ 18 years; 4. Failure of 2 of the following antidepressants, each used for ≥ 1 month, unless member’s age is ≥ 65, clinically significant adverse effects are experienced, or all are contraindicated: venlafaxine, fluoxetine, atomoxetine, clomipramine, protriptyline; 5. Dose does not exceed 35.6 mg (two 17.8 mg tablets) per day. Approval duration: Medicaid/HIM – 12 months Commercial – Length of Benefit B. Narcolepsy with Excessive Daytime Sleepiness (must meet all): 1. Diagnosis of narcolepsy with EDS; 2. Prescribed by or in consultation with a neurologist or sleep medicine specialist; 3. Age ≥ 18 years; 4. Failure of a 1-month trial of one of the following central nervous system (CNS) stimulants at up to maximally indicated doses, unless clinically significant adverse effects are experienced or all are contraindicated: amphetamine immediate-release (IR), amphetamine, dextroamphetamine IR, dextroamphetamine, methylphenidate IR; *Prior authorization may be required for CNS stimulants Page 1 of 6 CLINICAL POLICY Pitolisant 5. -

Chapter 2 Molecular Aspects of Histamine Receptors
VU Research Portal Shedding Light on the Histamine H3 Receptor Mocking, T.A.M. 2020 document version Publisher's PDF, also known as Version of record Link to publication in VU Research Portal citation for published version (APA) Mocking, T. A. M. (2020). Shedding Light on the Histamine H3 Receptor: Photopharmacology and bioluminescent assays to study GPCRs. General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal ? Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. E-mail address: [email protected] Download date: 06. Oct. 2021 Chapter 2 Molecular aspects of histamine receptors Histamine mediates a multitude of physiological effects in the human body by activating four histamine receptor subtypes. Histamine receptors have proven to be promising drug targets in the treatment of a variety of diseases, including hay fever, gastric ulcers, inflammatory and neuropathological diseases. In this chapter the molecular aspects of histamine receptors are described, including expression profile, intracellular signaling, and how histamine receptor activity can be attenuated by ligands targeting the histamine receptor binding sites. -

Stress Impairs 5-HT2A Receptor-Mediated Serotonergic Facilitation of GABA Release in Juvenile Rat Basolateral Amygdala
Neuropsychopharmacology (2009) 34, 410–423 & 2009 Nature Publishing Group All rights reserved 0893-133X/09 $32.00 www.neuropsychopharmacology.org Stress Impairs 5-HT2A Receptor-Mediated Serotonergic Facilitation of GABA Release in Juvenile Rat Basolateral Amygdala 1,2 1 3 4 1 ,1,2 Xiaolong Jiang , Guoqiang Xing , Chunhui Yang , Ajay Verma , Lei Zhang and He Li* 1 Department of Psychiatry, Center for the Study of Traumatic Stress, Uniformed Services University of the Health Sciences, Bethesda, MD, USA; 2 3 Neuroscience Program, Uniformed Services University of the Health Sciences, Bethesda, MD, USA; Section on Neuropathology, Clinical Brain 4 Disorders Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, MD, USA; Department of Neurology, Uniformed Services University of the Health Sciences, Bethesda, MD, USA The occurrence of stress and anxiety disorders has been closely associated with alterations of the amygdala GABAergic system. In these disorders, dysregulation of the serotonergic system, a very important modulator of the amygdala GABAergic system, is also well recognized. The present study, utilizing a learned helplessness stress rat model, was designed to determine whether stress is capable of altering serotonergic modulation of the amygdala GABAergic system. In control rats, administration of 5-HT or a-methyl-5-HT, a 5-HT2 receptor agonist, to basolateral amygdala (BLA) slices dramatically enhanced frequency and amplitude of spontaneous inhibitory postsynaptic currents (sIPSCs). This effect was blocked by selective 5-HT2A receptor antagonists while a selective 5-HT2B receptor agonist and a selective 5-HT2C receptor agonist were without effect on sIPSCs. Double immunofluorescence labeling demonstrated that the 5-HT2A receptor is primarily localized to parvalbumin-containing BLA interneurons. -

Histamine Receptors
Tocris Scientific Review Series Tocri-lu-2945 Histamine Receptors Iwan de Esch and Rob Leurs Introduction Leiden/Amsterdam Center for Drug Research (LACDR), Division Histamine is one of the aminergic neurotransmitters and plays of Medicinal Chemistry, Faculty of Sciences, Vrije Universiteit an important role in the regulation of several (patho)physiological Amsterdam, De Boelelaan 1083, 1081 HV, Amsterdam, The processes. In the mammalian brain histamine is synthesised in Netherlands restricted populations of neurons that are located in the tuberomammillary nucleus of the posterior hypothalamus.1 Dr. Iwan de Esch is an assistant professor and Prof. Rob Leurs is These neurons project diffusely to most cerebral areas and have full professor and head of the Division of Medicinal Chemistry of been implicated in several brain functions (e.g. sleep/ the Leiden/Amsterdam Center of Drug Research (LACDR), VU wakefulness, hormonal secretion, cardiovascular control, University Amsterdam, The Netherlands. Since the seventies, thermoregulation, food intake, and memory formation).2 In histamine receptor research has been one of the traditional peripheral tissues, histamine is stored in mast cells, eosinophils, themes of the division. Molecular understanding of ligand- basophils, enterochromaffin cells and probably also in some receptor interaction is obtained by combining pharmacology specific neurons. Mast cell histamine plays an important role in (signal transduction, proliferation), molecular biology, receptor the pathogenesis of various allergic conditions. After mast cell modelling and the synthesis and identification of new ligands. degranulation, release of histamine leads to various well-known symptoms of allergic conditions in the skin and the airway system. In 1937, Bovet and Staub discovered compounds that antagonise the effect of histamine on these allergic reactions.3 Ever since, there has been intense research devoted towards finding novel ligands with (anti-) histaminergic activity. -

Receptor Antagonist (H RA) Shortages | May 25, 2020 2 2 2 GERD4,5 • Take This Opportunity to Determine If Continued Treatment Is Necessary
H2-receptor antagonist (H2RA) Shortages Background . 2 H2RA Alternatives . 2 Therapeutic Alternatives . 2 Adults . 2 GERD . 3 PUD . 3 Pediatrics . 3 GERD . 3 PUD . 4 Tables Table 1: Health Canada–Approved Indications of H2RAs . 2 Table 2: Oral Adult Doses of H2RAs and PPIs for GERD . 4 Table 3: Oral Adult Doses of H2RAs and PPIs for PUD . 5 Table 4: Oral Pediatric Doses of H2RAs and PPIs for GERD . 6 Table 5: Oral Pediatric Doses of H2RAs and PPIs for PUD . 7 References . 8 H2-receptor antagonist (H2RA) Shortages | May 25, 2020 1 H2-receptor antagonist (H2RA) Shortages BACKGROUND Health Canada recalls1 and manufacturer supply disruptions may be causing shortages of commonly used acid-reducing medications called histamine H2-receptor antagonists (H2RAs) . H2RAs include cimetidine, famotidine, nizatidine and ranitidine . 2 There are several Health Canada–approved indications of H2RAs (see Table 1); this document addresses the most common: gastroesophageal reflux disease (GERD) and peptic ulcer disease (PUD) . 2 TABLE 1: HEALTH CANADA–APPROVED INDICATIONS OF H2RAs H -Receptor Antagonists (H RAs) Health Canada–Approved Indications 2 2 Cimetidine Famotidine Nizatidine Ranitidine Duodenal ulcer, treatment ü ü ü ü Duodenal ulcer, prophylaxis — ü ü ü Benign gastric ulcer, treatment ü ü ü ü Gastric ulcer, prophylaxis — — — ü GERD, treatment ü ü ü ü GERD, maintenance of remission — ü — — Gastric hypersecretion,* treatment ü ü — ü Self-medication of acid indigestion, treatment and prophylaxis — ü† — ü† Acid aspiration syndrome, prophylaxis — — — ü Hemorrhage from stress ulceration or recurrent bleeding, — — — ü prophylaxis ü = Health Canada–approved indication; GERD = gastroesophageal reflux disease *For example, Zollinger-Ellison syndrome . -

Tall Man Lettering List REPORT DECEMBER 2013 1
Tall Man Lettering List REPORT DECEMBER 2013 1 TALL MAN LETTERING LIST REPORT WWW.HQSC.GOVT.NZ Published in December 2013 by the Health Quality & Safety Commission. This document is available on the Health Quality & Safety Commission website, www.hqsc.govt.nz ISBN: 978-0-478-38555-7 (online) Citation: Health Quality & Safety Commission. 2013. Tall Man Lettering List Report. Wellington: Health Quality & Safety Commission. Crown copyright ©. This copyright work is licensed under the Creative Commons Attribution-No Derivative Works 3.0 New Zealand licence. In essence, you are free to copy and distribute the work (including other media and formats), as long as you attribute the work to the Health Quality & Safety Commission. The work must not be adapted and other licence terms must be abided. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nd/3.0/nz/ Copyright enquiries If you are in doubt as to whether a proposed use is covered by this licence, please contact: National Medication Safety Programme Team Health Quality & Safety Commission PO Box 25496 Wellington 6146 ACKNOWLEDGEMENTS The Health Quality & Safety Commission acknowledges the following for their assistance in producing the New Zealand Tall Man lettering list: • The Australian Commission on Safety and Quality in Health Care for advice and support in allowing its original work to be either reproduced in whole or altered in part for New Zealand as per its copyright1 • The Medication Safety and Quality Program of Clinical Excellence Commission, New South