Chapter 1 Chapter 1: the Gospel According to LUCA
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Spatio-Temporal Study of Microbiology in the Stratified Oxic-Hypoxic-Euxinic, Freshwater- To-Hypersaline Ursu Lake
Spatio-temporal insights into microbiology of the freshwater-to- hypersaline, oxic-hypoxic-euxinic waters of Ursu Lake Baricz, A., Chiriac, C. M., Andrei, A-., Bulzu, P-A., Levei, E. A., Cadar, O., Battes, K. P., Cîmpean, M., enila, M., Cristea, A., Muntean, V., Alexe, M., Coman, C., Szekeres, E. K., Sicora, C. I., Ionescu, A., Blain, D., O’Neill, W. K., Edwards, J., ... Banciu, H. L. (2020). Spatio-temporal insights into microbiology of the freshwater-to- hypersaline, oxic-hypoxic-euxinic waters of Ursu Lake. Environmental Microbiology. https://doi.org/10.1111/1462-2920.14909, https://doi.org/10.1111/1462-2920.14909 Published in: Environmental Microbiology Document Version: Peer reviewed version Queen's University Belfast - Research Portal: Link to publication record in Queen's University Belfast Research Portal Publisher rights Copyright 2019 Wiley. This work is made available online in accordance with the publisher’s policies. Please refer to any applicable terms of use of the publisher. General rights Copyright for the publications made accessible via the Queen's University Belfast Research Portal is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The Research Portal is Queen's institutional repository that provides access to Queen's research output. Every effort has been made to ensure that content in the Research Portal does not infringe any person's rights, or applicable UK laws. If you discover content in the Research Portal that you believe breaches copyright or violates any law, please contact [email protected]. -

The 7Th White Coat Ceremony at SSPPS by Binh Tran, Pharmd
The 7th White Coat Ceremony at SSPPS By Binh Tran, PharmD The White Coat Ceremony at the Skaggs School of Pharmacy and Pharmaceutical Sciences (SSPPS) on September 25th held special meaning as the University of California - San Diego celebrates its 50th Anniversary this year. SSPPS faculty, alumni, student pharmacists, the incoming class of 2014, their families and their friends listened to Dean Palmer Taylor’s welcoming address in the spacious Health Sciences auditorium modeled with panels made of eucalyptus wood. Dr Rita Shane, Director of Pharmacy Services at Cedars-Sinai Medical Center and Assistant Dean of Clinical Pharmacy at the UCSF School of Pharmacy delivered an energizing speech on The Power of Differentiation: Will you be a Brand or Generic? Attributes mentioned were competency, responsibility and accountability, intellectual curiosity, being able to seize opportunities, and establishing credibility. Dr. Anthony Manoguerra, Associate Dean for Student Affairs, reported that the class of 2014’s grade point average of 3.71 broke the record set by the previous class which averaged 3.70. The White Coat hooding by Dean Taylor was a moving ceremony with Justine Abella being last due to a recent foot injury. The class recited the Pharmacist’s Oath, which brought back memories of our own experiences. The Student Welcome by second- year student, Ammar Zanial, was heartfelt and reminiscent of his comments during pharmacy conference class. Little did I know at the time that the student pharmacist who volunteered at many outreaches in the community and helped backstage during Cultural Fusion Night was the President of his class. In closing, Dean Taylor commented on the ingeniousness of the class when he observed them tacitly move their seats in order to step closer to the stage. -

NC10 Phylum Anaerobs? Methanotrophs? Where Can I Find Them?
NC10 phylum Anaerobs? Methanotrophs? Where can I find them? Beate Kraft Microbial Diversity 2012 Abstract Methylomirabilis oxyfera, the only cultured member of the NC10 phylum performs the newly discovered pathway of NO-dismutation. In this study different habitats were screened for the presence of member of this phylum and the diversity of the sequences obtained was analyzed. Furthermore enrichment cultures for nitrite reduction coupled to methane oxidation were set up. Indeed, the presence of NC10 seems to be associated with nitrite and methane rich fresh water environments. Introduction Recently a new nitrite reduction pathway, NO-dismutation, coupled to methane oxidation has been discovered (Ettwig et al. 2010). In NO-dismutation nitrite is oxidized to NO as in denitrification but then NO dimutated into N2 and O2 instead of being further reduced to N2O. The generated oxygen is then used for methane oxidation. The organism responsible for this process is Methylomirabils oxfera. It is the only cultured member of the candidate phylum NC10. Fig 1: Pathway of NO-dismutation (from Ettwig et al. 2010) It remains open if other members of the NC10 phylum share the same metabolism and respiratory pathway or if they are metabolically more diverse. 16S sequences that fall into the NC10 phylum have been found in mainly oxygen limited freshwater habitats such as lakes including lake sediments, rice paddy soils, wastewater sludge and ditches (Deutzmann and Schink 2011, Luesken et a.l 2011). Their occurrence apparently seem to be correlated with the presence of methane and nitrite in the habitat. This would suggest a similar respiratory pathway as in Methylomirabilis oxyfera. -

Yu-Chen Ling and John W. Moreau
Microbial Distribution and Activity in a Coastal Acid Sulfate Soil System Introduction: Bioremediation in Yu-Chen Ling and John W. Moreau coastal acid sulfate soil systems Method A Coastal acid sulfate soil (CASS) systems were School of Earth Sciences, University of Melbourne, Melbourne, VIC 3010, Australia formed when people drained the coastal area Microbial distribution controlled by environmental parameters Microbial activity showed two patterns exposing the soil to the air. Drainage makes iron Microbial structures can be grouped into three zones based on the highest similarity between samples (Fig. 4). Abundant populations, such as Deltaproteobacteria, kept constant activity across tidal cycling, whereas rare sulfides oxidize and release acidity to the These three zones were consistent with their geological background (Fig. 5). Zone 1: Organic horizon, had the populations changed activity response to environmental variations. Activity = cDNA/DNA environment, low pH pore water further dissolved lowest pH value. Zone 2: surface tidal zone, was influenced the most by tidal activity. Zone 3: Sulfuric zone, Abundant populations: the heavy metals. The acidity and toxic metals then Method A Deltaproteobacteria Deltaproteobacteria this area got neutralized the most. contaminate coastal and nearby ecosystems and Method B 1.5 cause environmental problems, such as fish kills, 1.5 decreased rice yields, release of greenhouse gases, Chloroflexi and construction damage. In Australia, there is Gammaproteobacteria Gammaproteobacteria about a $10 billion “legacy” from acid sulfate soils, Chloroflexi even though Australia is only occupied by around 1.0 1.0 Cyanobacteria,@ Acidobacteria Acidobacteria Alphaproteobacteria 18% of the global acid sulfate soils. Chloroplast Zetaproteobacteria Rare populations: Alphaproteobacteria Method A log(RNA(%)+1) Zetaproteobacteria log(RNA(%)+1) Method C Method B 0.5 0.5 Cyanobacteria,@ Bacteroidetes Chloroplast Firmicutes Firmicutes Bacteroidetes Planctomycetes Planctomycetes Ac8nobacteria Fig. -

Are You an Invited Speaker? a Bibliometric Analysis of Elite Groups for Scholarly Events in Bioinformatics
Are You an Invited Speaker? A Bibliometric Analysis of Elite Groups for Scholarly Events in Bioinformatics Senator Jeong, Sungin Lee, and Hong-Gee Kim Biomedical Knowledge Engineering Laboratory, Seoul National University, 28–22 YeonGeon Dong, Jongno Gu, Seoul 110–749, Korea. E-mail: {senator, sunginlee, hgkim}@snu.ac.kr Participating in scholarly events (e.g., conferences, work- evaluation, but it would be hard to claim that they have pro- shops, etc.) as an elite-group member such as an orga- vided comprehensive lists of evaluation measurements. This nizing committee chair or member, program committee article aims not to provide such lists but to add to the current chair or member, session chair, invited speaker, or award winner is beneficial to a researcher’s career develop- practices an alternative metric that complements existing per- ment.The objective of this study is to investigate whether formance measures to give a more comprehensive picture of elite-group membership for scholarly events is represen- scholars’ performance. tative of scholars’ prominence, and which elite group is By one definition (Jeong, 2008), a scholarly event is the most prestigious. We collected data about 15 global “a sequentially and spatially organized collection of schol- (excluding regional) bioinformatics scholarly events held in 2007. We sampled (via stratified random sampling) ars’ interactions with the intention of delivering and shar- participants from elite groups in each event. Then, bib- ing knowledge, exchanging research ideas, and performing liometric indicators (total citations and h index) of seven related activities.” As such, scholarly events are communica- elite groups and a non-elite group, consisting of authors tion channels from which our new evaluation tool can draw who submitted at least one paper to an event but were its supporting evidence. -
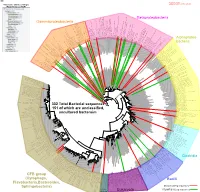
Bacteria Clostridia Bacilli Eukaryota CFB Group
AM935842.1.1361 uncultured Burkholderiales bacterium Class Betaproteobacteria AY283260.1.1552 Alcaligenes sp. PCNB−2 Class Betaproteobacteria AM934953.1.1374 uncultured Burkholderiales bacterium Class Betaproteobacteria AJ581593.1.1460 uncultured betaAM936569.1.1351 proteobacterium uncultured Class Betaproteobacteria Derxia sp. Class Betaproteobacteria AJ581621.1.1418 uncultured beta proteobacterium Class Betaproteobacteria DQ248272.1.1498 uncultured soil bacterium soil uncultured DQ248272.1.1498 DQ248235.1.1498 uncultured soil bacterium RS49 DQ248270.1.1496 uncultured soil bacterium DQ256489.1.1211 Variovorax paradoxus Class Betaproteobacteria Class paradoxus Variovorax DQ256489.1.1211 AF523053.1.1486 uncultured Comamonadaceae bacterium Class Betaproteobacteria AY706442.1.1396 uncultured bacterium uncultured AY706442.1.1396 AJ536763.1.1422 uncultured bacterium CS000359.1.1530 Variovorax paradoxus Class Betaproteobacteria Class paradoxus Variovorax CS000359.1.1530 AY168733.1.1411 uncultured bacterium AJ009470.1.1526 uncultured bacterium SJA−62 Class Betaproteobacteria Class SJA−62 bacterium uncultured AJ009470.1.1526 AY212561.1.1433 uncultured bacterium D16212.1.1457 Rhodoferax fermentans Class Betaproteobacteria Class fermentans Rhodoferax D16212.1.1457 AY957894.1.1546 uncultured bacterium AJ581620.1.1452 uncultured beta proteobacterium Class Betaproteobacteria RS76 AY625146.1.1498 uncultured bacterium RS65 DQ316832.1.1269 uncultured beta proteobacterium Class Betaproteobacteria DQ404909.1.1513 uncultured bacterium uncultured DQ404909.1.1513 AB021341.1.1466 bacterium rM6 AJ487020.1.1500 uncultured bacterium uncultured AJ487020.1.1500 RS7 RS86RC AF364862.1.1425 bacterium BA128 Class Betaproteobacteria AY957931.1.1529 uncultured bacterium uncultured AY957931.1.1529 CP000884.723807.725332 Delftia acidovorans SPH−1 Class Betaproteobacteria AY957923.1.1520 uncultured bacterium uncultured AY957923.1.1520 RS18 AY957918.1.1527 uncultured bacterium uncultured AY957918.1.1527 AY945883.1.1500 uncultured bacterium AF526940.1.1489 uncultured Ralstonia sp. -

The Phylogenetic Composition and Structure of Soil Microbial Communities Shifts in Response to Elevated Carbon Dioxide
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by University of Minnesota Digital Conservancy The ISME Journal (2012) 6, 259–272 & 2012 International Society for Microbial Ecology All rights reserved 1751-7362/12 www.nature.com/ismej ORIGINAL ARTICLE The phylogenetic composition and structure of soil microbial communities shifts in response to elevated carbon dioxide Zhili He1, Yvette Piceno2, Ye Deng1, Meiying Xu1,3, Zhenmei Lu1,4, Todd DeSantis2, Gary Andersen2, Sarah E Hobbie5, Peter B Reich6 and Jizhong Zhou1,2 1Institute for Environmental Genomics, Department of Botany and Microbiology, University of Oklahoma, Norman, OK, USA; 2Ecology Department, Earth Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA, USA; 3Guangdong Provincial Key Laboratory of Microbial Culture Collection and Application, Guangdong Institute of Microbiology, Guangzhou, China; 4College of Life Sciences, Zhejiang University, Hangzhou, China; 5Department of Ecology, Evolution, and Behavior, St Paul, MN, USA and 6Department of Forest Resources, University of Minnesota, St Paul, MN, USA One of the major factors associated with global change is the ever-increasing concentration of atmospheric CO2. Although the stimulating effects of elevated CO2 (eCO2) on plant growth and primary productivity have been established, its impacts on the diversity and function of soil microbial communities are poorly understood. In this study, phylogenetic microarrays (PhyloChip) were used to comprehensively survey the richness, composition and structure of soil microbial communities in a grassland experiment subjected to two CO2 conditions (ambient, 368 p.p.m., versus elevated, 560 p.p.m.) for 10 years. The richness based on the detected number of operational taxonomic units (OTUs) significantly decreased under eCO2. -

Changes in the Human Skin Microbiome Over One Year's Time
American Journal of Microbiology 3 (2): 18-30, 2012 ISSN 1948-982x © 2012 Science Publications doi:10.3844/ajmsp.2012.18.30 Published Online 3 (2) 2012 (http://www.thescipub.com/ajm.toc) Changes in the Human Skin Microbiome Over One Year’s Time 1,2 Richard W. Hyman, 1Farbod Babrzadeh, 1Curtis Palm, 1Chunlin Wang, 1Marilyn Fukushima and 1,2,3 Ronald W. Davis 1Stanford Genome Technology Center, 855 S California Avenue, Palo Alto, CA 94304, USA 2Department of Biochemistry, 3Department of Genetics, Stanford University College of Medicine, Stanford, CA 94305, USA ABSTRACT Human skin comprises a large number of distinguishable ecological niches. To describe fully the human skin microbiome, it will be necessary to identify the bacteria in each niche and to distinguish the commensal bacteria from the temporary residents. To contribute to the description of the human skin microbiome and employing a gene-based technology, we have identified the bacteria in two niches: the front and back of the base of the neck and over the course of one year. There were 50 volunteers and a total of 232 neck skin swabs. Roche 454 Tag pyrosequencing was employed to sequence a short hypervariable sequence region (V6) of the 16S ribosomal RNA gene. To identify the bacteria corresponding to the front and back of the neck for each volunteer, the “Classifier” software in the “Pyrosequencing” section of the Ribosomal Database Project was employed. The bacteria on virtually all 232 neck skin swabs were classified into bacterial Class. The skin microbiome of these two niches was composed principally of a mixture of five Classes of bacteria: Actinobacteria , Alphaproteobacteria , Bacilli , Betaproteobacteria and Gammaproteobacteria . -

Your Scientific Reputation Class Slides
Your Scientific Reputation and Being a Responsible Member of Society Peter D Sottile, MD October 2020 • Silence personal devices. • Stay muted when not talking. • Set up in a quiet location. Housekeeping: Zoom • Remain attentive. Avoid checking Etiquette: email/phone/web. • Use the Chat function to ask questions or get technical help. • Use your full name, not an alias. Receiving credit for attendance: To satisfy the NIH Requirement for Instruction in the Responsible Conduct of Research, the following are required in order to receive credit for attendance: Attend the full 90 minutes of the training. Attending any 8 out of the 9 RCR seminars we offer will satisfy the NIH requirement. Keep your video camera on throughout the session. NIH requirements for RCR training specify face- to-face discussion. Participate interactively throughout the session. Participate in discussions, respond to polls, and sign the attendance sheet (link will be distributed in the Chat). Raise your Hand to participate in discussions: In order to participate in discussions, raise your hand. Try it now! • Click “Participants” at the bottom of your screen. • Click “Raise Hand” in the popup window. • Click “Lower Hand” to stop raising your hand. 3 Objectives: • Explain why a good scientific reputation is important in academia • Describe the factors that contribute to a good scientific reputation • Identify the components to being a responsible member of the scientific community • Describe threats that may harm one’s scientific reputation, and responses to challenges -

Annual Scientific Report 2011 Annual Scientific Report 2011 Designed and Produced by Pickeringhutchins Ltd
European Bioinformatics Institute EMBL-EBI Annual Scientific Report 2011 Annual Scientific Report 2011 Designed and Produced by PickeringHutchins Ltd www.pickeringhutchins.com EMBL member states: Austria, Croatia, Denmark, Finland, France, Germany, Greece, Iceland, Ireland, Israel, Italy, Luxembourg, the Netherlands, Norway, Portugal, Spain, Sweden, Switzerland, United Kingdom. Associate member state: Australia EMBL-EBI is a part of the European Molecular Biology Laboratory (EMBL) EMBL-EBI EMBL-EBI EMBL-EBI EMBL-European Bioinformatics Institute Wellcome Trust Genome Campus, Hinxton Cambridge CB10 1SD United Kingdom Tel. +44 (0)1223 494 444, Fax +44 (0)1223 494 468 www.ebi.ac.uk EMBL Heidelberg Meyerhofstraße 1 69117 Heidelberg Germany Tel. +49 (0)6221 3870, Fax +49 (0)6221 387 8306 www.embl.org [email protected] EMBL Grenoble 6, rue Jules Horowitz, BP181 38042 Grenoble, Cedex 9 France Tel. +33 (0)476 20 7269, Fax +33 (0)476 20 2199 EMBL Hamburg c/o DESY Notkestraße 85 22603 Hamburg Germany Tel. +49 (0)4089 902 110, Fax +49 (0)4089 902 149 EMBL Monterotondo Adriano Buzzati-Traverso Campus Via Ramarini, 32 00015 Monterotondo (Rome) Italy Tel. +39 (0)6900 91402, Fax +39 (0)6900 91406 © 2012 EMBL-European Bioinformatics Institute All texts written by EBI-EMBL Group and Team Leaders. This publication was produced by the EBI’s Outreach and Training Programme. Contents Introduction Foreword 2 Major Achievements 2011 4 Services Rolf Apweiler and Ewan Birney: Protein and nucleotide data 10 Guy Cochrane: The European Nucleotide Archive 14 Paul Flicek: -

An Abstract of the Thesis Of
AN ABSTRACT OF THE THESIS OF Kaitlin Mae McConnell, for the degree of Master of Science in Microbiology presented on September 6, 2019. Title: Reef Microbiomes of Mo’orea, French Polynesia, a Socio-Ecological Island System Abstract approved: ______________________________________________________ Andrew R. Thurber At a time of rapid global change, a socio-ecological system (SES) approach can provide a framework through which to quantify and communicate the risks and uncertainties of coupled human-natural systems. Islands, and tropical coral reef islands in particular, can be excellent models for SES research since they may be considered as both closed and open systems, where agents-- both human and non- human-- and their interactions, may be considered at a variety of spatial and temporal scales and with diverse levels of complexity. Bacteria and Archaea communities are the executants of biochemical cycling in systems, and alterations in the structure of microbial communities can provide indications of system-wide metabolic shifts driven by natural and human forces. In this thesis, I first present patterns of spatial and temporal variability in microbial communities across a tropical coral island reefscape, where I hypothesized: Microbiome host, habitat and sampling location will house distinct microbiomes (beta diversity) that differ between seasons. Microbiomes were primarily host specific, and host specificity varied with structural scale: microbiomes of reef sediment, seawater and corals were different from each other. Zooming in, the three species of corals examined also had distinct microbial communities associated with them. These host- specific microbiomes varied differentially across spatial scale, such as island side, reef habitat (fore, back and fringing reefs) and sampling locations. -

Microbial Community of a Gasworks Aquifer and Identification of Nitrate
Water Research 132 (2018) 146e157 Contents lists available at ScienceDirect Water Research journal homepage: www.elsevier.com/locate/watres Microbial community of a gasworks aquifer and identification of nitrate-reducing Azoarcus and Georgfuchsia as key players in BTEX degradation * Martin Sperfeld a, Charlotte Rauschenbach b, Gabriele Diekert a, Sandra Studenik a, a Institute of Microbiology, Friedrich Schiller University Jena, Department of Applied and Ecological Microbiology, Philosophenweg 12, 07743 Jena, Germany ® b JENA-GEOS -Ingenieurbüro GmbH, Saalbahnhofstraße 25c, 07743 Jena, Germany article info abstract Article history: We analyzed a coal tar polluted aquifer of a former gasworks site in Thuringia (Germany) for the Received 9 August 2017 presence and function of aromatic compound-degrading bacteria (ACDB) by 16S rRNA Illumina Received in revised form sequencing, bamA clone library sequencing and cultivation attempts. The relative abundance of ACDB 18 December 2017 was highest close to the source of contamination. Up to 44% of total 16S rRNA sequences were affiliated Accepted 18 December 2017 to ACDB including genera such as Azoarcus, Georgfuchsia, Rhodoferax, Sulfuritalea (all Betaproteobacteria) Available online 20 December 2017 and Pelotomaculum (Firmicutes). Sequencing of bamA, a functional gene marker for the anaerobic benzoyl-CoA pathway, allowed further insights into electron-accepting processes in the aquifer: bamA Keywords: Environmental pollutions sequences of mainly nitrate-reducing Betaproteobacteria were abundant in all groundwater samples, Microbial communities whereas an additional sulfate-reducing and/or fermenting microbial community (Deltaproteobacteria, Bioremediation Firmicutes) was restricted to a highly contaminated, sulfate-depleted groundwater sampling well. By Box pathway conducting growth experiments with groundwater as inoculum and nitrate as electron acceptor, or- Functional gene marker ganisms related to Azoarcus spp.