Download This Article PDF Format
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Downloads/DL Praevention/Fachwissen/Gefahrstoffe/TOXIKOLOGI SCHE BEWERTUNGEN/Bewertungen/Toxbew072-L.Pdf
Distribution Agreement In presenting this thesis or dissertation as a partial fulfillment of the requirements for an advanced degree from Emory University, I hereby grant to Emory University and its agents the non-exclusive license to archive, make accessible, and display my thesis or dissertation in whole or in part in all forms of media, now or hereafter known, including display on the world wide web. I understand that I may select some access restrictions as part of the online submission of this thesis or dissertation. I retain all ownership rights to the copyright of the thesis or dissertation. I also retain the right to use in future works (such as articles or books) all or part of this thesis or dissertation. Signature: _____________________________ ______________ Jedidiah Samuel Snyder Date Statistical analysis of concentration-time extrapolation factors for acute inhalation exposures to hazardous substances By Jedidiah S. Snyder Master of Public Health Global Environmental Health _________________________________________ P. Barry Ryan, Ph.D. Committee Chair _________________________________________ Eugene Demchuk, Ph.D. Committee Member _________________________________________ Paige Tolbert, Ph.D. Committee Member Statistical analysis of concentration-time extrapolation factors for acute inhalation exposures to hazardous substances By Jedidiah S. Snyder Bachelor of Science in Engineering, B.S.E. The University of Iowa 2010 Thesis Committee Chair: P. Barry Ryan, Ph.D. An abstract of A thesis submitted to the Faculty of the Rollins School of Public Health of Emory University in partial fulfillment of the requirements for the degree of Master of Public Health in Global Environmental Health 2015 Abstract Statistical analysis of concentration-time extrapolation factors for acute inhalation exposures to hazardous substances By Jedidiah S. -

Synthesis of Isothiocyanates Using DMT/NMM/Tso− As a New Desulfurization Reagent
molecules Article Synthesis of Isothiocyanates Using DMT/NMM/TsO− as a New Desulfurization Reagent Łukasz Janczewski 1,* , Dorota Kr˛egiel 2 and Beata Kolesi ´nska 1 1 Faculty of Chemistry, Institute of Organic Chemistry, Lodz University of Technology, Zeromskiego 116, 90-924 Lodz, Poland; [email protected] 2 Department of Environmental Biotechnology, Faculty of Biotechnology and Food Sciences, Lodz University of Technology, Wolczanska 171/173, 90-924 Lodz, Poland; [email protected] * Correspondence: [email protected] Abstract: Thirty-three alkyl and aryl isothiocyanates, as well as isothiocyanate derivatives from esters of coded amino acids and from esters of unnatural amino acids (6-aminocaproic, 4-(aminomethyl)benzoic, and tranexamic acids), were synthesized with satisfactory or very good yields (25–97%). Synthesis was performed in a “one-pot”, two-step procedure, in the presence of organic base (Et3N, DBU or NMM), and carbon disulfide via dithiocarbamates, with 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4- methylmorpholinium toluene-4-sulfonate (DMT/NMM/TsO−) as a desulfurization reagent. For the synthesis of aliphatic and aromatic isothiocyanates, reactions were carried out in a microwave reactor, and selected alkyl isothiocyanates were also synthesized in aqueous medium with high yields (72–96%). Isothiocyanate derivatives of L- and D-amino acid methyl esters were synthesized, under conditions without microwave radiation assistance, with low racemization (er 99 > 1), and their absolute configuration was confirmed by circular dichroism. Isothiocyanate derivatives of natural and unnatural amino acids were evaluated for antibacterial activity on E. coli and S. aureus bacterial strains, where the Citation: Janczewski, Ł.; Kr˛egiel,D.; most active was ITC 9e. -

Argonne Report.Pdf
CONTENTS NOTATION ........................................................................................................................... xi ABSTRACT ........................................................................................................................... 1 1 INTRODUCTION ........................................................................................................... 5 1.1 Overview of the Emergency Response Guidebook ................................................ 5 1.2 Organization of this Report ..................................................................................... 7 2 GENERAL METHODOLOGY ....................................................................................... 9 2.1 TIH List ................................................................................................................... 10 2.1.1 Background ................................................................................................. 10 2.1.2 Changes in the TIH List for the ERG2012 ................................................. 11 2.2 Shipment and Release Scenarios ............................................................................ 11 2.2.1 Shipment Profiles ........................................................................................ 12 2.2.2 Treatment of Chemical Agents ................................................................... 14 2.3 Generics, Mixtures, and Solutions .......................................................................... 17 2.4 Analysis of Water-Reactive -

NAC/AEGL Committee Meeting Minutes, December 2007
National Advisory Committee (NAC) for Acute Exposure Guideline Levels (AEGLs) for Hazardous Substances December 5-7, 2007 Meeting-44 Highlights Orlando World Center Marriott 8701 World Center Drive Orlando, FL INTRODUCTION Ernest Falke distributed a CD containing the most recent Technical Support Documents for all AEGL program chemicals that are proposed, interim, or final status. Dr. Falke also stated that the Prepublication Copy of NAS Volume 6 was released in September, 2007, and that the published volume should be available for distribution by the March meeting. Paul Tobin informed the group that the NAC/AEGL committee was recognized by the FACA awards program twice (last year and this year). There are 27 EPA FACAs, and only the NAC/AEGL was recognized twice; a total of only 3 EPA FACAs were recognized. Specifically, the NAC/AEGL was recognized for timeliness of Federal Register notices, transparency of the process, public input (both via the meeting and the Federal Register), and international impact. The draft NAC/AEGL-43 meeting highlights were reviewed. Due to a rounding error, the 30-minute AEGL-3 value for dichlorosilanes should be 110 ppm, not 105 ppm. A motion was made by Bob Benson and seconded by John Hinz to accept the minutes as proposed with the aforementioned correction. The motion passed unanimously by a show of hands (Appendix A). The Final NAC/AEGL-43 meeting highlights are attached (Appendix B). The highlights of the NAC/AEGL-44 meeting are summarized below along with the Meeting Agenda (Attachment 1) and the Attendee List (Attachment 2). The subject categories of the highlights do not necessarily follow the order listed in the NAC/AEGL-44 Agenda. -

2020 Emergency Response Guidebook
2020 A guidebook intended for use by first responders A guidebook intended for use by first responders during the initial phase of a transportation incident during the initial phase of a transportation incident involving hazardous materials/dangerous goods involving hazardous materials/dangerous goods EMERGENCY RESPONSE GUIDEBOOK THIS DOCUMENT SHOULD NOT BE USED TO DETERMINE COMPLIANCE WITH THE HAZARDOUS MATERIALS/ DANGEROUS GOODS REGULATIONS OR 2020 TO CREATE WORKER SAFETY DOCUMENTS EMERGENCY RESPONSE FOR SPECIFIC CHEMICALS GUIDEBOOK NOT FOR SALE This document is intended for distribution free of charge to Public Safety Organizations by the US Department of Transportation and Transport Canada. This copy may not be resold by commercial distributors. https://www.phmsa.dot.gov/hazmat https://www.tc.gc.ca/TDG http://www.sct.gob.mx SHIPPING PAPERS (DOCUMENTS) 24-HOUR EMERGENCY RESPONSE TELEPHONE NUMBERS For the purpose of this guidebook, shipping documents and shipping papers are synonymous. CANADA Shipping papers provide vital information regarding the hazardous materials/dangerous goods to 1. CANUTEC initiate protective actions. A consolidated version of the information found on shipping papers may 1-888-CANUTEC (226-8832) or 613-996-6666 * be found as follows: *666 (STAR 666) cellular (in Canada only) • Road – kept in the cab of a motor vehicle • Rail – kept in possession of a crew member UNITED STATES • Aviation – kept in possession of the pilot or aircraft employees • Marine – kept in a holder on the bridge of a vessel 1. CHEMTREC 1-800-424-9300 Information provided: (in the U.S., Canada and the U.S. Virgin Islands) • 4-digit identification number, UN or NA (go to yellow pages) For calls originating elsewhere: 703-527-3887 * • Proper shipping name (go to blue pages) • Hazard class or division number of material 2. -

EXPOSURE and INTERACTION the Potential Health Impacts of Using Multiple Pesticides
EXPOSURE AND INTERACTION The Potential Health Impacts of Using Multiple Pesticides Virginia Zaunbrecher, J.D. Dale Hattis, Ph.D. Ron Melnick, Ph.D. Susan Kegley, Ph.D. Timothy Malloy, J.D. John Froines, Ph.D. Authors Virginia Zaunbrecher, J.D., UCLA Sustainable Technology & Policy Program Dale Hattis, Ph.D., Clark University Ron Melnick, Ph.D., Ron Melnick Consulting Susan Kegley, Ph.D., Pesticide Research Institute Timothy Malloy, J.D., UCLA Sustainable Technology & Policy Program John Froines, Ph.D., UCLA Sustainable Technology & Policy Program Contributors Lindsey Sears, M.A. Timothy J. Brown, Ph.D. Camille Sears Christina Batteate, M.P.H., UCLA Sustainable Technology & Policy Program Charlene Nguyen, UCLA Sustainable Technology & Policy Program UCLA Sustainable Technology & Policy Program The Sustainable Technology & Policy Program, a joint undertaking of the UCLA School of Law and the Fielding School of Public Health, is an interdisciplinary research and education program. Its mission is to advance public health and environmental protection by integrating science, law, and engineering to promote prevention in public and private decision making. Acknowledgements This research was made possible by generous support from the Clarence E. Heller Charitable Foundation. The Center for Occupational and Environmental Health (COEH) at UCLA provided additional support. The authors bear responsibility for any factual errors. Recommendations and views expressed are those of the authors and do not necessarily represent those of the funder or of the University of California. Appendices and Supplementary Materials All appendices and supplementary materials mentioned in this report are available at: http://www.stpp.ucla.edu/node/586 KEY TERMS AND DEFINITIONS Combined exposure to a single stressor (e.g. -

Particularly Hazardous Substances Human Reproductive Page 1 of 25 March 2011 Carcinogen Hazard
Particularly Hazardous Substances Human Reproductive Page 1 of 25 March 2011 Carcinogen Hazard CAS Number order Acutely Probable Reactive Female Known Toxic Male CAS Fetal Number Chemical 00–00–1 Nickel compounds 00–00–2 Chromium [VI] Compunds 00–00–3 Cadmium compounds 00–00–4 Chlorophenols (polychlorophenols) 00–00–5 Hexachlorocyclohexanes 00–00–6 Lead compounds, inorganic 00–00–7 Methyl and other organic mercury compounds 00–01–0 Polychlorinated biphenyls (PCBs) ‐ all forms 00–01–1 Organolithium compounds 00–01–3 Botulinum Toxins 00–01–4 Clostridium perfringens, epsilon toxin 00–01–5 Conotoxins 00–01–6 Ricin isolates 00–01–7 Saxitoxins 00–01–8 Staphylococcal enterotoxins 00–01–9 Tetrodotoxins 00–02–0 Tricothecene mycotoxins 50–00–0 Formaldehyde (Formalin) (Paraformaldeyde) 50–06–6 Phenobarbital 50–07–7 Mitomycin C 50–18–0 Cyclophosphamide 50–29–3 DDT [p,p'‐DDT] 50–32–8 Benzo[a]pyrene 50–35–1 Thalidomide 50–55–5 Reserpine 51–21–8 5‐Fluorouracil 51–28–5 Dinitrophenol Particularly Hazardous Substances Human Reproductive Page 2 of 25 March 2011 Carcinogen Hazard CAS Number order Acutely Probable Reactive Female Known Toxic Male CAS Fetal Number Chemical 51–52–5 Propylthiouracil 51–75–2 HN2 (nitrogen mustard‐2) 51–79–6 Ethyl carbamate (Urethane) 52–24–4 Thiotepa 52–67–5 Valine, 3‐mercapto‐, D‐ 53–70–3 Dibenz [a,h]anthracene 53–96–3 2‐acetylaminofluorene 54–62–6 Aminopterin 55–18–5 N‐Nitrosodiethylamine 55–63–0 Nitrogycerine 55–86–7 Nitrogen Mustard Hydrochloride 55–98–1 1,4‐Butanediol dimethanesulfonate (Busulfan; Myleran) 56–04–2 Methylthiouracil -

Irritant Compounds: Military Respiratory Irritants. Part I
Mil. Med. Sci. Lett. (Voj. Zdrav. Listy) 2015, vol. 84(3), p. 128-139 ISSN 0372-7025 DOI: 10.31482/mmsl.2015.014 REVIEW ARTICLE IRRITANT COMPOUNDS: MILITARY RESPIRATORY IRRITANTS. PART I. LACRIMATORS Jiri Patocka 1,3 , Kamil Kuca 2,3 1 Department of Radiology and Toxicology, Faculty of Health and Social Studies, University of South Bohemia, Ceske Budejovice, Czech Republic 2 Center of Advanced Studies, Faculty of Military Health Sciences, University of Defence, Hradec Kralove, Czech Republic 3 Biomedical Research Centre, University Hospital; Hradec Kralove, Czech Republic Received 29 th September 2014. Revised 24 th May 2015. Published 4 th September 2015. Summary World War I was a conflict where chemical warfare was first used on a massive scale. The earliest chemical attack occurred on the Western Front in October 1914 in Neuve Chapelle, but its effects were so minimal that the Allies learned about it only after the war from German documents. The attack in the Bolimow area, carried out by the Germans against the Russian army with artillery shells containing gas T (xylyl and benzyl bromides), was therefore the first attack on a massive scale recorded on the victim side. The attack, which occurred after it, made it possible to obtain some tactical success, but without a strategic breakthrough. Some of the later German attacks on the Eastern Front where chlorine was used proved to be more effective, but despite many victims there was not any major strategic success achieved. The Russians did not take attempts to use chemical weapons in the World War I. Key words: respiratory irritants; irritant gases; chemical warfare agents; riot control agents; World War I INTRODUCTION comfort to acute airway and lung injury and even death. -

TIH/PIH List
Hazardous Materials Designated as TIH/PIH (consolidated AAR and Railinc lists) 3/12/2007 STCC Proper Shipping Name 4921402 2-CHLOROETHANAL 4921495 2-METHYL-2-HEPTANETHIOL 4921741 3,5-DICHLORO-2,4,6-TRIFLUOROPYRIDINE 4921401 ACETONE CYANOHYDRIN, STABILIZED 4927007 ACROLEIN, STABILIZED 4921019 ALLYL ALCOHOL 4923113 ALLYL CHLOROFORMATE 4921004 ALLYLAMINE 4904211 AMMONIA SOLUTION 4920360 AMMONIA SOLUTIONS 4904209 AMMONIA, ANHYDROUS 4904210 AMMONIA, ANHYDROUS 4904879 AMMONIA, ANHYDROUS 4920359 AMMONIA, ANHYDROUS 4923209 ARSENIC TRICHLORIDE 4920135 ARSINE 4932010 BORON TRIBROMIDE 4920349 BORON TRICHLORIDE 4920522 BORON TRIFLUORIDE 4936110 BROMINE 4920715 BROMINE CHLORIDE 4918505 BROMINE PENTAFLUORIDE 4936106 BROMINE SOLUTIONS 4918507 BROMINE TRIFLUORIDE 4921727 BROMOACETONE 4920343 CARBON MONOXIDE AND HYDROGEN MIXTURE, COMPRESSED 4920399 CARBON MONOXIDE, COMPRESSED 4920511 CARBON MONOXIDE, REFRIGERATED LIQUID 4920559 CARBONYL FLUORIDE 4920351 CARBONYL SULFIDE 4920523 CHLORINE 4920189 CHLORINE PENTAFLUORIDE 4920352 CHLORINE TRIFLUORIDE 4921558 CHLOROACETONE, STABILIZED 4921009 CHLOROACETONITRILE 4923117 CHLOROACETYL CHLORIDE 4921414 CHLOROPICRIN 4920516 CHLOROPICRIN AND METHYL BROMIDE MIXTURES 4920547 CHLOROPICRIN AND METHYL BROMIDE MIXTURES 4920392 CHLOROPICRIN AND METHYL CHLORIDE MIXTURES 4921746 CHLOROPIVALOYL CHLORIDE 4930204 CHLOROSULFONIC ACID 4920527 COAL GAS, COMPRESSED 4920102 COMMPRESSED GAS, TOXIC, FLAMMABLE, CORROSIVE, N.O.S. 4920303 COMMPRESSED GAS, TOXIC, FLAMMABLE, CORROSIVE, N.O.S. 4920304 COMMPRESSED GAS, TOXIC, FLAMMABLE, CORROSIVE, N.O.S. 4920305 COMMPRESSED GAS, TOXIC, FLAMMABLE, CORROSIVE, N.O.S. 4920101 COMPRESSED GAS, TOXIC, CORROSIVE, N.O.S. 4920300 COMPRESSED GAS, TOXIC, CORROSIVE, N.O.S. 4920301 COMPRESSED GAS, TOXIC, CORROSIVE, N.O.S. 4920324 COMPRESSED GAS, TOXIC, CORROSIVE, N.O.S. 4920331 COMPRESSED GAS, TOXIC, CORROSIVE, N.O.S. 4920165 COMPRESSED GAS, TOXIC, FLAMMABLE, N.O.S. 4920378 COMPRESSED GAS, TOXIC, FLAMMABLE, N.O.S. 4920379 COMPRESSED GAS, TOXIC, FLAMMABLE, N.O.S. -
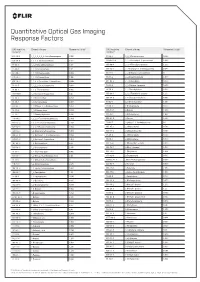
Quantitative Optical Gas Imaging Response Factors
Quantitative Optical Gas Imaging Response Factors CAS registry Chemical name Response factor† CAS registry Chemical name Response factor† number* number* 431-89-0 1_1_1_2_3_3_3-Heptafluoropropane 0.103 75-89-8 2_2_2-Trifluoroethanol 0.326 630-20-6 1_1_1_2-Tetrachloroethane 0.132 25256-77-4 2_2_4-Trimethyl-1_3-pentanediol 1.086 75-68-3 1_1_1-Chlorodifluoroethane 0.117 107-40-4 2_2_4-Trimethyl-2-pentene 1.262 71-55-6 1_1_1-Trichloroethane 0.121 306-83-2 2_2-Dichloro-1_1_1-trifluoroethane 0.047 421-50-1 1_1_1-Trifluoroacetone 0.086 76-11-9 2_2-Difluorotetrachloroethane 0 420-46-2 1_1_1-Trifluoroethane 0.106 75-83-2 2_2-Dimethyl_butane 1.944 354-14-3 1_1_2_2-Tetrachloro-1-fluoroethane 0.046 431-03-8 2_3-Butanedione 0.274 79-34-5 1_1_2_2-Tetrachloroethane 0.069 78-88-6 2_3-Dichloro-1-propene 0.279 79-00-5 1_1_2-Trichloroethane 0.102 79-29-8 2_3-Dimethylbutane 1.014 1717-00-6 1_1-Dichloro-1-fluoroethane 0.12 107-39-1 2_4_4-Trimethyl-1-pentene 1.212 75-34-3 1_1-Dichloroethane 0.272 584-84-9 2_4-Diisocyanatetoluene 0.895 75-35-4 1_1-Dichloroethene 0.017 87-62-7 2_6-Dimethylaniline 1.308 471-43-2 1_1-Difluoro-2_2-dichloroethane 0.352 75-26-3 2-Bromopropane 0.733 73-37-6 1_1-Difluoroethane 0.535 107-01-7 2-Butene 1.204 57-14-7 1_1-Dimethylhydrazine 0.897 111-76-2 2-Butoxyethanol 1.962 119-64-2 1_2_3_4-Tetrahydronaphthalene 2.439 554-61-0 2-Carene 1.286 488-23-3 1_2_3_4-Tetramethylbenzene 1.484 75-88-7 2-Chloro-1_1_1-trifluoroethane 0.1 527-53-7 1_2_3_5-Tetramethylbenzene 1.402 107-07-3 2-Chloroethanol 0.593 124-73-2 1_2-Dibomotetrafluoroethane -

Association of American Railroads
ASSOCIATION OF AMERICAN RAILROADS K.B. Dorsey Executive Director - Tank Car Safety April 30, 2021 Circular No. OT-55-R (CPC-1384) Subject: Recommended Railroad Operating Practices for Transportation of Hazardous Materials TO: MEMBERS AND PRIVATE CAR OWNERS AAR’s Safety and Operations department is amending OT-55 by adding back in Appendix 3, the list of Environmentally Sensitive Chemicals. The Sample Request for Hazardous Materials Commodity Flow Information will now be in Appendix 4. These are the only changes from OT-55-Q. AAR Circular No. OT-55-R (attached) becomes effective April 30, 2021 and supersedes OT-55-Q. Under the provisions of Standard S-050, which may be found on the TTCI web site (www.AAR.com), this circular reflects the final action on this matter. Respectfully Submitted, K.B. Dorsey 425 Third Street SW, Suite 1000 | Washington, DC 20024 | P (202) 639-2262 | F (202) 639-2439 [email protected] | www.aar.org ASSOCIATION OF AMERICAN RAILROADS Circular OT-55-R Effective April 30, 2021 Recommended Railroad Operating Practices For Transportation of Hazardous Materials Road Operating Practices I. "Key Trains" A. Definition: A “Key Train” is any train with: • One tank car load of Poison or Toxic Inhalation Hazard1 (PIH or TIH) (Hazard Zone A, B, C, or D), anhydrous ammonia (UN1005), or ammonia solutions (UN3318), or; • 20 car loads or intermodal portable tank loads of any combination of hazardous material, or; • One or more car loads of Spent Nuclear Fuel (SNF), High Level Radioactive Waste (HLRW). Appendices A and B are the lists of PIH/TIH sorted by Hazard Class and alphabetically proper shipping name, and by Hazard Class and Hazardous Materials Response Code (HMRC), respectively, Appendix 1 is a list of SNF and HLRW with 49 Hazmat Codes, Appendix 2 is a list of time sensitive materials and Appendix 3 is a form for requesting hazardous materials commodity flow information. -
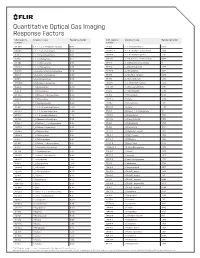
Quantitative Optical Gas Imaging Response Factors CAS Registry Chemical Name Response Factor† CAS Registry Chemical Name Response Factor† Number* Number*
Quantitative Optical Gas Imaging Response Factors CAS registry Chemical name Response factor† CAS registry Chemical name Response factor† number* number* 431-89-0 1_1_1_2_3_3_3-Heptafluoropropane 0.103 75-89-8 2_2_2-Trifluoroethanol 0.326 630-20-6 1_1_1_2-Tetrachloroethane 0.132 25256-77-4 2_2_4-Trimethyl-1_3-pentanediol 1.086 75-68-3 1_1_1-Chlorodifluoroethane 0.117 107-40-4 2_2_4-Trimethyl-2-pentene 1.262 71-55-6 1_1_1-Trichloroethane 0.121 306-83-2 2_2-Dichloro-1_1_1-trifluoroethane 0.047 421-50-1 1_1_1-Trifluoroacetone 0.086 76-11-9 2_2-Difluorotetrachloroethane 0 420-46-2 1_1_1-Trifluoroethane 0.106 75-83-2 2_2-Dimethyl_butane 1.944 354-14-3 1_1_2_2-Tetrachloro-1-fluoroethane 0.046 431-03-8 2_3-Butanedione 0.274 79-34-5 1_1_2_2-Tetrachloroethane 0.069 78-88-6 2_3-Dichloro-1-propene 0.279 79-00-5 1_1_2-Trichloroethane 0.102 79-29-8 2_3-Dimethylbutane 1.014 1717-00-6 1_1-Dichloro-1-fluoroethane 0.12 107-39-1 2_4_4-Trimethyl-1-pentene 1.212 75-34-3 1_1-Dichloroethane 0.272 584-84-9 2_4-Diisocyanatetoluene 0.895 75-35-4 1_1-Dichloroethene 0.017 87-62-7 2_6-Dimethylaniline 1.308 471-43-2 1_1-Difluoro-2_2-dichloroethane 0.352 75-26-3 2-Bromopropane 0.733 73-37-6 1_1-Difluoroethane 0.535 107-01-7 2-Butene 1.204 57-14-7 1_1-Dimethylhydrazine 0.897 111-76-2 2-Butoxyethanol 1.962 119-64-2 1_2_3_4-Tetrahydronaphthalene 2.439 554-61-0 2-Carene 1.286 488-23-3 1_2_3_4-Tetramethylbenzene 1.484 75-88-7 2-Chloro-1_1_1-trifluoroethane 0.1 527-53-7 1_2_3_5-Tetramethylbenzene 1.402 107-07-3 2-Chloroethanol 0.593 124-73-2 1_2-Dibomotetrafluoroethane