Genes Potentially Associated with Familial Hypercholesterolemia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Evaluation of the Role of STAP1 in Familial Hypercholesterolemia Magdalena Danyel1,2, Claus-Eric Ott 2, Thomas Grenkowitz1, Bastian Salewsky1, Andrew A
www.nature.com/scientificreports OPEN Evaluation of the role of STAP1 in Familial Hypercholesterolemia Magdalena Danyel1,2, Claus-Eric Ott 2, Thomas Grenkowitz1, Bastian Salewsky1, Andrew A. Hicks 3, Christian Fuchsberger3, Elisabeth Steinhagen-Thiessen1, 1 1 1,4 Received: 17 January 2019 Thomas Bobbert , Ursula Kassner & Ilja Demuth Accepted: 2 August 2019 Familial hypercholesterolemia (FH) is characterised by elevated serum levels of low-density lipoprotein Published: xx xx xxxx cholesterol (LDL-C) and a substantial risk for cardiovascular disease. The autosomal-dominant FH is mostly caused by mutations in LDLR (low density lipoprotein receptor), APOB (apolipoprotein B), and PCSK9 (proprotein convertase subtilisin/kexin). Recently, STAP1 has been suggested as a fourth causative gene. We analyzed STAP1 in 75 hypercholesterolemic patients from Berlin, Germany, who are negative for mutations in canonical FH genes. In 10 patients with negative family history, we additionally screened for disease causing variants in LDLRAP1 (low density lipoprotein receptor adaptor protein 1), associated with autosomal-recessive hypercholesterolemia. We identifed one STAP1 variant predicted to be disease causing. To evaluate association of serum lipid levels and STAP1 carrier status, we analyzed 20 individuals from a population based cohort, the Cooperative Health Research in South Tyrol (CHRIS) study, carrying rare STAP1 variants. Out of the same cohort we randomly selected 100 non-carriers as control. In the Berlin FH cohort STAP1 variants were rare. In the CHRIS cohort, we obtained no statistically signifcant diferences between carriers and non-carriers of STAP1 variants with respect to lipid traits. Until such an association has been verifed in more individuals with genetic variants in STAP1, we cannot estimate whether STAP1 generally is a causative gene for FH. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Redefining the Specificity of Phosphoinositide-Binding by Human
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.20.163253; this version posted June 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Redefining the specificity of phosphoinositide-binding by human PH domain-containing proteins Nilmani Singh1†, Adriana Reyes-Ordoñez1†, Michael A. Compagnone1, Jesus F. Moreno Castillo1, Benjamin J. Leslie2, Taekjip Ha2,3,4,5, Jie Chen1* 1Department of Cell & Developmental Biology, University of Illinois at Urbana-Champaign, Urbana, IL 61801; 2Department of Biophysics and Biophysical Chemistry, Johns Hopkins University School of Medicine, Baltimore, MD 21205; 3Department of Biophysics, Johns Hopkins University, Baltimore, MD 21218; 4Department of Biomedical Engineering, Johns Hopkins University, Baltimore, MD 21205; 5Howard Hughes Medical Institute, Baltimore, MD 21205, USA †These authors contributed equally to this work. *Correspondence: [email protected]. bioRxiv preprint doi: https://doi.org/10.1101/2020.06.20.163253; this version posted June 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. ABSTRACT Pleckstrin homology (PH) domains are presumed to bind phosphoinositides (PIPs), but specific interaction with and regulation by PIPs for most PH domain-containing proteins are unclear. Here we employed a single-molecule pulldown assay to study interactions of lipid vesicles with full-length proteins in mammalian whole cell lysates. -

The Crucial Roles of Apolipoproteins E and C-III in Apob Lipoprotein Metabolism in Normolipidemia and Hypertriglyceridemia
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Harvard University - DASH The crucial roles of apolipoproteins E and C-III in apoB lipoprotein metabolism in normolipidemia and hypertriglyceridemia The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Sacks, Frank M. 2015. “The Crucial Roles of Apolipoproteins E and C-III in apoB Lipoprotein Metabolism in Normolipidemia and Hypertriglyceridemia.” Current Opinion in Lipidology 26 (1) (February): 56–63. doi:10.1097/mol.0000000000000146. Published Version doi:10.1097/MOL.0000000000000146 Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:30203554 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Open Access Policy Articles, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#OAP HHS Public Access Author manuscript Author Manuscript Author ManuscriptCurr Opin Author Manuscript Lipidol. Author Author Manuscript manuscript; available in PMC 2016 February 01. Published in final edited form as: Curr Opin Lipidol. 2015 February ; 26(1): 56–63. doi:10.1097/MOL.0000000000000146. The crucial roles of apolipoproteins E and C-III in apoB lipoprotein metabolism in normolipidemia and hypertriglyceridemia Frank M. Sacks Department of Nutrition, Harvard School of Public Health, Boston, Massachusetts, USA Abstract Purpose of review—To describe the roles of apolipoprotein C-III (apoC-III) and apoE in VLDL and LDL metabolism Recent findings—ApoC-III can block clearance from the circulation of apolipoprotein B (apoB) lipoproteins, whereas apoE mediates their clearance. -

Coexpression of ATP-Binding Cassette Proteins ABCG5 and ABCG8 Permits Their Transport to the Apical Surface
Coexpression of ATP-binding cassette proteins ABCG5 and ABCG8 permits their transport to the apical surface Gregory A. Graf, … , Jonathan C. Cohen, Helen H. Hobbs J Clin Invest. 2002;110(5):659-669. https://doi.org/10.1172/JCI16000. Article Cardiology Mutations in either ATP-binding cassette (ABC) G5 or ABCG8 cause sitosterolemia, an autosomal recessive disorder of sterol trafficking. To determine the site of action of ABCG5 and ABCG8, we expressed recombinant, epitope-tagged mouse ABCG5 and ABCG8 in cultured cells. Both ABCG5 and ABCG8 underwent N-linked glycosylation. When either protein was expressed individually in cells, the N-linked sugars remained sensitive to Endoglycosidase H (Endo H). When ABCG5 and ABCG8 were coexpressed, the attached sugars were Endo H–resistant and neuraminidase-sensitive, indicating that the proteins were transported to the trans-Golgi complex. The mature, glycosylated forms of ABCG5 and ABCG8 coimmunoprecipitated, consistent with heterodimerization of these two proteins. The Endo H–sensitive forms of ABCG5 and ABCG8 were confined to the endoplasmic reticulum (ER), whereas the mature forms were present in non-ER fractions in cultured hepatocytes. Immunoelectron microscopy revealed ABCG5 and ABCG8 on the plasma membrane of these cells. In polarized WIF-B cells, recombinant ABCG5 localized to the apical (canalicular) membrane when coexpressed with ABCG8, but not when expressed alone. To our knowledge this is the first direct demonstration that trafficking of an ABC half-transporter to the cell surface requires the presence of its dimerization partner. Find the latest version: https://jci.me/16000/pdf Coexpression of ATP-binding cassette See the related Commentary beginning on page 605. -

Transcriptome-Wide Profiling of Multiple RNA Modifications Simultaneously at Single-Base Resolution,” by Vahid Khoddami, Archana Yerra, Timothy L
Correction BIOCHEMISTRY, CHEMISTRY Correction for “Transcriptome-wide profiling of multiple RNA modifications simultaneously at single-base resolution,” by Vahid Khoddami, Archana Yerra, Timothy L. Mosbruger, Aaron M. Fleming, Cynthia J. Burrows, and Bradley R. Cairns, which was first published March 14, 2019; 10.1073/pnas.1817334116 (Proc Natl Acad Sci USA 116:6784–6789). The authors note that the following statement should be added to the Acknowledgments: “This work was also supported by Grant R01 GM093099 from the NIH/National Institute of General Medical Sciences (to C.J.B.).” Published under the PNAS license. Published online April 22, 2019. www.pnas.org/cgi/doi/10.1073/pnas.1905628116 9136 | PNAS | April 30, 2019 | vol. 116 | no. 18 www.pnas.org Downloaded by guest on October 2, 2021 Transcriptome-wide profiling of multiple RNA modifications simultaneously at single-base resolution Vahid Khoddamia,b,c,1,2, Archana Yerrab,c,1, Timothy L. Mosbrugerd, Aaron M. Fleminge, Cynthia J. Burrowse,3, and Bradley R. Cairnsb,c,3 aDepartment of Cell Biology, Harvard Medical School, Boston, MA 02115; bHoward Hughes Medical Institute, University of Utah School of Medicine, Salt Lake City, UT 84112; cDepartment of Oncological Sciences, Huntsman Cancer Institute, University of Utah School of Medicine, Salt Lake City, UT 84112; dBioinformatics Shared Resource, Huntsman Cancer Institute, University of Utah School of Medicine, Salt Lake City, UT 84112; and eDepartment of Chemistry, University of Utah, Salt Lake City, UT 84112 Contributed by Cynthia J. Burrows, January 25, 2019 (sent for review October 9, 2018; reviewed by Juan D. Alfonzo, Thomas Carell, and Peter C. -

Mai Muudatuntuu Ti on Man Mini
MAIMUUDATUNTUU US009809854B2 TI ON MAN MINI (12 ) United States Patent ( 10 ) Patent No. : US 9 ,809 ,854 B2 Crow et al. (45 ) Date of Patent : Nov . 7 , 2017 Whitehead et al. (2005 ) Variation in tissue - specific gene expression ( 54 ) BIOMARKERS FOR DISEASE ACTIVITY among natural populations. Genome Biology, 6 :R13 . * AND CLINICAL MANIFESTATIONS Villanueva et al. ( 2011 ) Netting Neutrophils Induce Endothelial SYSTEMIC LUPUS ERYTHEMATOSUS Damage , Infiltrate Tissues, and Expose Immunostimulatory Mol ecules in Systemic Lupus Erythematosus . The Journal of Immunol @(71 ) Applicant: NEW YORK SOCIETY FOR THE ogy , 187 : 538 - 552 . * RUPTURED AND CRIPPLED Bijl et al. (2001 ) Fas expression on peripheral blood lymphocytes in MAINTAINING THE HOSPITAL , systemic lupus erythematosus ( SLE ) : relation to lymphocyte acti vation and disease activity . Lupus, 10 :866 - 872 . * New York , NY (US ) Crow et al . (2003 ) Microarray analysis of gene expression in lupus. Arthritis Research and Therapy , 5 :279 - 287 . * @(72 ) Inventors : Mary K . Crow , New York , NY (US ) ; Baechler et al . ( 2003 ) Interferon - inducible gene expression signa Mikhail Olferiev , Mount Kisco , NY ture in peripheral blood cells of patients with severe lupus . PNAS , (US ) 100 ( 5 ) : 2610 - 2615. * GeneCards database entry for IFIT3 ( obtained from < http : / /www . ( 73 ) Assignee : NEW YORK SOCIETY FOR THE genecards. org /cgi - bin / carddisp .pl ? gene = IFIT3 > on May 26 , 2016 , RUPTURED AND CRIPPLED 15 pages ) . * Navarra et al. (2011 ) Efficacy and safety of belimumab in patients MAINTAINING THE HOSPITAL with active systemic lupus erythematosus : a randomised , placebo FOR SPECIAL SURGERY , New controlled , phase 3 trial . The Lancet , 377 :721 - 731. * York , NY (US ) Abramson et al . ( 1983 ) Arthritis Rheum . -

Cardiovascular Disease Products
Cardiovascular Disease Products For more information, visit: www.bosterbio.com Cardiovascular Disease Research Cardiovascular disease is the leading cause of death in developed nations. Boster Bio aims to supply researchers with high-quality antibodies and ELISA kits so they can make new discoveries and help save lives. In this catalogue you will find a comprehensive list of high-affinity Boster antibodies and high sensitivity Boster ELISA kits targeted at proteins associated with cardiovascular disease. Boster: The Fastest Growing About Bosterbio Antibody Company In 2015 Boster is an antibody manufacturer founded in 1993 by histologist Steven Xia. Over the past two decades, Boster and its products have been cited in over 20,000 publications and counting. The firm specializes in developing antibodies and ELISA kits that feature high affinity, Boster Bio received the CitaAb award for high specificity at affordable the greatest increase in number of prices. citations during 2015 than any other antibody manufacturer. Table of Contents Boster Cardiovascular Disease Related Antibodies…………..………..... 2 Boster Cardiovascular Disease Related ELISA Kits……………………..…. 9 1 High Affinity Boster Antibodies Boster supplies only the highest quality antibodies. Our high-affinity polyclonal and monoclonal antibodies are thoroughly validated by Western Blotting, Immunohistochemistry and ELISA. This is our comprehensive catalog of our antibody products related to cardiovascular disease, sorted in alphabetical order by target gene name. Catalog No Product Name -

Apolipoproteins and Their Association with Cardiometabolic Risk
Nutr Hosp. 2015;32(6):2674-2683 ISSN 0212-1611 • CODEN NUHOEQ S.V.R. 318 Original / Síndrome metabólico Apolipoproteins and their association with cardiometabolic risk biomarkers in adolescents Mellina Neyla de Lima Albuquerque1, Alcides da Silva Diniz1 and Ilma Kruze Grande de Arruda1 1Department of Nutrition. Federal University of Pernambuco, Recife, Brazil. Abstract APOLIPOPROTEÍNAS Y SU ASOCIACIÓN CON BIOMARCADORES DE RIESGO Introduction: the apoB/apo A-I ratio has been reported CARDIOMETABÓLICO EN ADOLESCENTES as an important predictor of cardiovascular risk, being superior to lipids, lipoproteins and conventional lipid ra- tios. Resumen Objective: to investigate the association between apo- Introducción: la razón apo B/apo A-I sigue siendo lipoproteins A-I and B, and the apolipoprotein B/apoli- reportada como un predictor importante de riesgo car- poprotein A-I ratio and cardiometabolic risk variables in diovascular, superior a lípidos, lipoproteínas y razones adolescents. lipídicas convencionales. Objetivo: investigar la asocia- Methods: this was a cross-sectional study including ción entre las apolipoproteínas A-I y B y la razón apoli- 104 adolescents of public schools in Recife during the poproteína B/apolipoproteína A-I con variables de riesgo months of March/April, 2013. Sociodemographic, an- cardiometabólico en adolescentes. thropometric, clinical and biochemical variables were Métodos: estudio de corte transversal que incluye a analysed. The apolipoproteins were analysed via Immu- 104 adolescentes de escuelas públicas de -
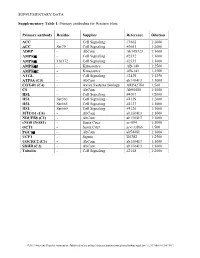
Supplementary Table 1
SUPPLEMENTARY DATA Supplementary Table 1. Primary antibodies for Western blots. Primary antibody Residue Supplier Reference Dilution ACC - Cell Signaling #3662 1:2000 ACC Ser79 Cell Signaling #3661 1:2000 ADRP - AbCam Ab108323 1:1000 AMPKα - Cell Signaling #2532 1:1000 AMPKα Thr172 Cell Signaling #2535 1:1000 AMPKα1 - Kinasource AB-140 1:2500 AMPKα2 - Kinasource AB-141 1:2500 ATGL - Cell Signaling #2439 1:1250 ATP5A (C5) - AbCam ab110413 1:1000 COX4l1 (C4) - Aviva Systems Biology ARP42784 1:500 CS - AbCam Ab96600 1:1000 HSL - Cell Signaling #4107 1:2000 HSL Ser563 Cell Signaling #4139 1:2000 HSL Ser565 Cell Signaling #4137 1:1000 HSL Ser660 Cell Signaling #4126 1:1000 MTCO1 (C4) - AbCam ab110413 1:1000 NDUFB8 (C1) - AbCam ab110413 1:1000 eNOS (NOS3) - Santa Cruz sc-654 1:1000 OCT1 - Santa Cruz sc-133866 1:500 PGC1α - AbCam ab54481 1:1000 UCP1 - Sigma U6382 1:2500 UQCRC2 (C3) - AbCam ab110413 1:1000 SDHB (C2) - AbCam ab110413 1:1000 Tubulin - Cell Signaling #2148 1:2000 ©2013 American Diabetes Association. Published online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db13-0194/-/DC1 SUPPLEMENTARY DATA Supplementary Table 2. Primer sequences for qRT-PCR. Gene Accession number Forward primer Reverse primer Abca1 NM_013454.3 CCCAGAGCAAAAAGCGACTC GGTCATCATCACTTTGGTCCTTG Abcg5 NM_031884 TGTCCTACAGCGTCAGCAACC GGCCACTCTCGATGTACAAGG Abcg8 NM_026180 TCCTGTGAGCTGGGCATCCGA CCCGCAGCCTGAGCTCCCTAT Acaca NM_133360.2 CAGCTGGTGCAGAGGTACCG TCTACTCGCAGGTACTGCCG Acacb NM_133904.2 GCGCCTACTATGAGGCCCAGCA ACAAACTCGGCTGGGGACGC Acly NM_134037.2 -

Endosomal PI(3)P Regulation by the COMMD/CCDC22/CCDC93
ARTICLE https://doi.org/10.1038/s41467-019-12221-6 OPEN Endosomal PI(3)P regulation by the COMMD/ CCDC22/CCDC93 (CCC) complex controls membrane protein recycling Amika Singla 1,5, Alina Fedoseienko2,5, Sai S.P. Giridharan3, Brittany L. Overlee2, Adam Lopez1, Da Jia 4, Jie Song1, Kayci Huff-Hardy1, Lois Weisman3, Ezra Burstein 1,6 & Daniel D. Billadeau2,6 1234567890():,; Protein recycling through the endolysosomal system relies on molecular assemblies that interact with cargo proteins, membranes, and effector molecules. Among them, the COMMD/CCDC22/CCDC93 (CCC) complex plays a critical role in recycling events. While CCC is closely associated with retriever, a cargo recognition complex, its mechanism of action remains unexplained. Herein we show that CCC and retriever are closely linked through sharing a common subunit (VPS35L), yet the integrity of CCC, but not retriever, is required to maintain normal endosomal levels of phosphatidylinositol-3-phosphate (PI(3)P). CCC complex depletion leads to elevated PI(3)P levels, enhanced recruitment and activation of WASH (an actin nucleation promoting factor), excess endosomal F-actin and trapping of internalized receptors. Mechanistically, we find that CCC regulates the phosphorylation and endosomal recruitment of the PI(3)P phosphatase MTMR2. Taken together, we show that the regulation of PI(3)P levels by the CCC complex is critical to protein recycling in the endosomal compartment. 1 Department of Internal Medicine, and Department of Molecular Biology, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA. 2 Division of Oncology Research and Department of Biochemistry and Molecular Biology, College of Medicine, Mayo Clinic, Rochester, MN 55905, USA. -

Apolipoprotein B
Laboratory Procedure Manual Analyte: Apolipoprotein B Matrix: Serum Method: Turbidimetric Assay on Roche Cobas® 6000 Method No.: Revised: As performed by: University of Minnesota – Advanced Research Diagnostics Laboratory (ARDL) Contact: Dr. Anthony Killeen, MD, PhD University of Minnesota Medical Center Fairview-University Medical Center University Campus Minneapolis, Minnesota Important Information for Users University of Minnesota – Advanced Research Diagnostics Laboratory (ARDL) periodically refines these laboratory methods. It is the responsibility of the user to contact the person listed on the title page of each write-up before using the analytical method to find out whether any changes have been made and what revisions, if any, have been incorporated. Apolipoprotein B NHANES 2015-2016 Public Release Data Set Information This document details the Lab Protocol for testing the items listed in the following table: Data File Name Variable Name SAS Label LBXAPB Apolipoprotein B (mg/dL) APOB_I LBDAPBSI Apolipoprotein B (g/L) 2 of 20 Apolipoprotein B NHANES 2015-2016 1. SUMMARY OF TEST PRINCIPLE AND CLINICAL RELEVANCE A. Clinical Relevance Apolipoproteins are the protein constituents of the lipoproteins. Apolipoprotein B (Apo B) is the major protein component of low-density lipoprotein (LDL). About one-third of the LDL particles provide cholesterol to peripheral cells. The other two-thirds are metabolized by the liver. LDL-uptake in all of these cells occurs via LDL receptors. Apo B levels increase in hypercholesterolemia, pregnancy, LDL receptor defects, bile obstruction and nephrotic syndrome. Apo B levels decrease in liver disease, sepsis and estrogen administration. The combined measurement of apolipoprotein A-1 (Apo A1, present in HDL) and Apo B and the calculation of the Apo B:Apo A1 ratio can reflect a lipid metabolism disorder and the risk of developing atherosclerosis or coronary heart disease.