The Role of Human Spliceosomal Component Srm300 in Neurodegenerative Disease
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Neural Mechanisms of Oxytocin and Serotonin Interaction in Non-Human Primates and Patients with Autism Arthur Lefevre
Neural mechanisms of oxytocin and serotonin interaction in non-human primates and patients with autism Arthur Lefevre To cite this version: Arthur Lefevre. Neural mechanisms of oxytocin and serotonin interaction in non-human primates and patients with autism. Neuroscience. Université de Lyon, 2016. English. NNT : 2016LYSE1323. tel-01508642 HAL Id: tel-01508642 https://tel.archives-ouvertes.fr/tel-01508642 Submitted on 14 Apr 2017 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. N°d’ordre NNT : xxx THESE de DOCTORAT DE L’UNIVERSITE DE LYON opérée au sein de l’Université Claude Bernard Lyon 1 Ecole Doctorale 476 (Neuroscience et Cognition) Spécialité de doctorat : Discipline : Neurosciences Soutenue publiquement le 13 Décembre 2016, par : Arthur LEFEVRE __________________________________________ Neural mechanisms of oxytocin and serotonin interaction in non- human primates and patients with autism __________________________________________ Thèse dirigée par Angela Sirigu (DRCE, CNRS) Devant le jury composé de : GERVAIS Rémi, Président, (Professeur à l’Université Lyon 1 Claude Bernard) CHINI BICE, Rapporteur, (Senior researcher at the Institute of Neuroscience, Milan) CHAKRABARTI Bhismadev, Rapporteur, (Associate Professor at university of Reading and Senior researcher at Cambridge university) KRAUS Christoph, Examinateur, (Senior researcher at the Medical university of Vienna) SIRIGU Angela, Directrice de thèse, (DRCE, Université de Lyon, CNRS) 1 UNIVERSITE CLAUDE BERNARD - LYON 1 Président de l’Université M. -

Section of Neurosurgery, Department of Surgery, University of Michigan Hospital, Ann Arbor, Michigan
ANATOMIC PATHWAYS RELATED TO PAIN IN FACE AND NECK* JAMES A. TAREN, M.D., AND EDGAR A. KAHN, M.D. Section of Neurosurgery, Department of Surgery, University of Michigan Hospital, Ann Arbor, Michigan (Received for publication May 19, 1961) HE remarkable ability of the higher V is most dorsal in the tract. Fig. 4 is a dia- portions of the central nervous system gram of the medulla 6 mm. below the obex T to readjust to surgical lesions presup- which we believe to be the optimal level for poses the existence of alternate pathways. tractotomy. The operation is done with the This hypothesis has been tested with regard patient in the sitting position to facilitate to a specific localized sensory input, pain exposure. The medullary incision, 4-5 mm. from the face. in depth, extends from the bulbar accessory Our method has been to study the degen- rootlet to a line extrapolated from the pos- eration of nerve fibers by Weil and Marchi terior rootlets of the ~ud cervical nerve root. techniques following various surgical lesions An adequate incision results in complete in man and monkey. The lesions have con- analgesia of all 8 divisions as well as analgesia sisted of total and selective retrogasserian in the distribution of VII, IX, and X except rhizotomy in 3 humans and 3 monkeys, for sparing of the vermilion border of the lips medullary tractotomy in ~ humans and (Fig. 5). A degree of ataxia of the ipsilateral monkeys, and extirpation of the cervical upper extremity usually accompanies effec- portion of the nucleus of the descending tract tive tractotomy and is caused by compromise of V in ~ monkeys. -

Dopamine and Serotonin Metabolism in Parkinsonian Models
Dopamine and Serotonin Metabolism in Parkinsonian Models Carmen de la Fuente Barrigon UCL Great Ormond Street Institute of Child Health Thesis submitted for the degree of Doctor of Philosophy (PhD) awarded by University College London (UCL). Funded by Marie Skłodowska-Curie Actions of the European Union’s Seventh Framework Programme (FP7). APRIL 2018 1 I, Carmen de la Fuente Barrigon confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. Signed ………………………………………….. Date ……………………………………………… 3 Dedico esta tesis a los pilares de mi vida. A mis padres, Tomás y Marisa. A mi hermana, Paloma. A mi amor, Francesco. Por vuestros sacrificios, paciencia, amor y apoyo incondicionales que me hacen ser quien soy hoy. 5 Abstract Parkinson’s disease (PD) is a neurodegenerative disorder caused by loss of dopaminergic neurons in the substantia nigra. Different pathogenic mechanisms have been implicated, including loss of mitochondrial complex I function and dysfunction of lysosomal glucocerebrosidase (GBA1) (Neumann et al., 2009; Schapira et al., 1990). Also, it has been hypothesised that serotonin metabolism could be affected in these patients due to the number of enzymes shared by both pathways (Albizu et al., 2011). This thesis considers the potential involvement of complex I and GBA1 in PD using HPLC analysis of changes in the extracellular levels of the metabolites of dopamine and serotonin, and the expression and activity of the enzymes of the dopamine pathway. Using SH-SY5Y cells, complex I deficiency was modelled using rotenone, and GBA1 deficiency was modelled using conduritol B epoxide (CBE). -

DR. Sanaa Alshaarawy
By DR. Sanaa Alshaarawy 1 By the end of the lecture, students will be able to : Distinguish the internal structure of the components of the brain stem in different levels and the specific criteria of each level. 1. Medulla oblongata (closed, mid and open medulla) 2. Pons (caudal, mid “Trigeminal level” and rostral). 3. Mid brain ( superior and inferior colliculi). Describe the Reticular formation (structure, function and pathway) being an important content of the brain stem. 2 1. Traversed by the Central Canal. Motor Decussation*. Spinal Nucleus of Trigeminal (Trigeminal sensory nucleus)* : ➢ It is a larger sensory T.S of Caudal part of M.O. nucleus. ➢ It is the brain stem continuation of the Substantia Gelatinosa of spinal cord 3 The Nucleus Extends : Through the whole length of the brain stem and upper segments of spinal cord. It lies in all levels of M.O, medial to the spinal tract of the trigeminal. It receives pain and temperature from face, forehead. Its tract present in all levels of M.O. is formed of descending fibers that terminate in the trigeminal nucleus. 4 It is Motor Decussation. Formed by pyramidal fibers, (75-90%) cross to the opposite side They descend in the Decuss- = crossing lateral white column of the spinal cord as the lateral corticospinal tract. The uncrossed fibers form the ventral corticospinal tract. 5 Traversed by Central Canal. Larger size Gracile & Cuneate nuclei, concerned with proprioceptive deep sensations of the body. Axons of Gracile & Cuneate nuclei form the internal arcuate fibers; decussating forming Sensory Decussation. Pyramids are prominent ventrally. 6 Formed by the crossed internal arcuate fibers Medial Leminiscus: Composed of the ascending internal arcuate fibers after their crossing. -

Neurophysiological and Neurohumoral Changes by High Frequency Stimulation (HFS) of Nucleus Accumbens (Nac)
From the Department of Neurosurgery of the University of Luebeck Director: Prof. Dr. med. Volker Tronnier Neurophysiological and neurohumoral changes by High Frequency Stimulation (HFS) of Nucleus Accumbens (NAc) Dissertation for Fulfillment of Requirements for the Doctoral Degree of the University of Luebeck from the Department of Natural Sciences Submitted by Ramya Varatharajan Thirugnanasambantham Born in Veeramanipatti-TN, India Luebeck, 2015 I Ramya Varatharajan Thirugnanasambantham Department of Neurosurgery University of Luebeck Ratzeburger allee 160 23562, Luebeck Germany First referee: Prof. Dr. Volker Tronnier Second referee: Prof. Dr. Thomas Martinetz Date of oral examination: 23.09.2015 Approved for printing, Luebeck: 30.09.2015 II Acknowledgements The hard work of so many people is involved in successful completion of this thesis, that a genuine list is virtually impossible. I convey my thanks and gratitude to Prof. Dr. med. Volker Tronnier, Department of Neurosurgery, for providing this opportunity to work under his guidance with this interesting topic. I express my deepest gratitude to Prof. Dr. med. Andreas Moser and Prof. Dr. rer. nat. Ulrich G. Hofmann for providing Laboratory Infrastructure, financial and moral support. Beyond scientific supervision, I was lucky to have the people from liquorlab, guiding me through all matters during my stay in Luebeck. I am thankful to Katharina Schnackenberg whose excellent experience in the laboratory and generous practical help was essential for the successful work at the lab. Thanks to Karin Wiegers, Marie Luis Reher and Gisa Brunk for being like friends and taking good care of me. I am very much thankful to Krishnakumar, Kevin Joseph, Sonya Neto, Mehrnaz Hazrati, Yijing Xie and Susanne Loeffler for their support, encouragement and timely suggestions. -

Imaging Elevated Brain Arachidonic Acid Signaling in Unanesthetized Serotonin Transporter (5-HTT)-Deficient Mice
Neuropsychopharmacology (2009) 34, 1695–1709 & 2009 Nature Publishing Group All rights reserved 0893-133X/09 $32.00 www.neuropsychopharmacology.org Imaging Elevated Brain Arachidonic Acid Signaling in Unanesthetized Serotonin Transporter (5-HTT)-Deficient Mice Mireille Basselin*,1, Meredith A Fox2, Lisa Chang1, Jane M Bell1, Dede Greenstein3, Mei Chen1, 2 1 Dennis L Murphy and Stanley I Rapoport 1Brain Physiology and Metabolism Section, National Institute on Aging, National Institutes of Health, Bethesda, MD, USA; 2Laboratory of Clinical Science, National Institutes of Health, Bethesda, MD, USA; 3Child Psychiatry Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, MD, USA Certain polymorphisms reduce serotonin (5-HT) reuptake transporter (5-HTT) function and increase susceptibility to psychiatric +/À disorders. Heterozygous (5-HTT )-deficient mice, models for humans with these polymorphisms, have elevated brain 5-HT concentrations and behavioral abnormalities. As postsynaptic 5-HT2A/2C receptors are coupled to cytosolic phospholipase A2 (cPLA2), which releases arachidonic acid (AA) from membrane phospholipid, 5-HTT-deficient mice may have altered brain AA signaling and metabolism. To test this hypothesis, signaling was imaged as an AA incorporation coefficient k* in unanesthetized homozygous knockout À/À +/À +/+ (5-HTT ), 5-HTT and wild-type (5-HTT ), mice following saline (baseline) or 1.5 mg/kg s.c. DOI, a partial 5-HT2A/2C receptor agonist. Enzyme activities, metabolite concentrations, and head-twitch responses to DOI were also measured. Baseline k* was widely +/À À/À +/+ +/+ elevated by 20–70% in brains of 5-HTT and 5-HTT compared to 5-HTT mice. DOI increased k* in 5-HTT mice, but decreased k* in 5-HTT-deficient mice. -
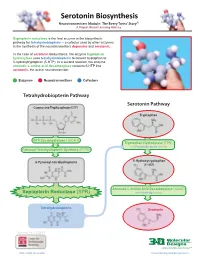
Serotonin Biosynthesis Neurotransmitters Module: the Beery Twins’ Story© a Project-Based Learning Activity
Serotonin Biosynthesis Neurotransmitters Module: The Beery Twins’ Story© A Project-Based Learning Activity Sepiapterin reductase is the final enzyme in the biosynthetic pathway for tetrahydrobiopterin – a cofactor used by other enzymes in the synthesis of the neurotransmitters dopamine and serotonin. In the case of serotonin biosynthesis, the enzyme tryptophan hydroxylase uses tetrahydrobiopterin to convert tryptophan to 5-hydroxytryptophan (5-HTP). In a second reaction, the enzyme aromatic L-amino acid decarboxylase converts 5-HTP into serotonin, the active neurotransmitter. Enzymes Neurotransmitters Cofactors Tetrahydrobiopterin Pathway Serotonin Pathway Guanosine Triphosphate (GTP) Tryptophan GTP Cyclohydrolase I (GCH1) Tryptophan Hydroxylase (TPH) with Tetrahydrobiopterin Cofactor Pyruvoyl-Tetrahydropterin Synthase (PTPS) 6-Pyruvoyl-tetrahydropterin 5-Hydroxytryptophan (5-HTP) Aromatic L-Amino Acid Decarboxylase (AAAD) Sepiapterin Reductase (SPR) with Vitamin B6 Cofactor Tetrahydrobiopterin Serotonin Version 1.4 -12/2015 ...where molecules become real TM http://cbm.msoe.edu www.3dmoleculardesigns.com Serotonin Biosynthesis Model Guide Neurotransmitters Module: The Beery Twins’ Story© A Project-Based Learning Activity TPH AAAD Tryptophan 5-Hydroxytryptophan Serotonin Tryptophan (Trp or W) is one of the 20 standard 5-Hydroxytryptophan, an intermediate The nal step in the serotonin biosynthesis amino acids and is an essential amino acid that molecule in the serotonin biosynthesis pathway requires the removal of the cannot be synthesized by the human body. pathway, is formed by the addition of a carboxylic acid group (COOH) from the Tryptophan is composed of the standard amino hydroxyl (OH) group to the fth carbon of the backbone of 5-hydroxytryptophan to form acid backbone with an indole ring side chain. indole ring of tryptophan. -

Lecture (6) Internal Structures of the Brainstem.Pdf
Internal structures of the Brainstem Neuroanatomy block-Anatomy-Lecture 6 Editing file Objectives At the end of the lecture, students should be able to: ● Distinguish the internal structure of the components of the brain stem in different levels and the specific criteria of each level. 1. Medulla oblongata (closed, mid and open medulla) 2. Pons (caudal and rostral). 3. Midbrain ( superior and inferior colliculi). Color guide ● Only in boys slides in Green ● Only in girls slides in Purple ● important in Red ● Notes in Grey Medulla oblongata Caudal (Closed) Medulla Traversed by the central canal Motor decussation (decussation of the pyramids) ● Formed by pyramidal fibers, (75-90%) cross to the opposite side ● They descend in the lateral white column of the spinal cord as the lateral corticospinal tract. ● The uncrossed fibers form the ventral corticospinal tract Trigeminal sensory nucleus. ● it is the larger sensory nucleus. ● The Nucleus Extends Through the whole length of the brainstem and its note :All CN V afferent sensory information enters continuation of the substantia gelatinosa of the spinal cord. the brainstem through the nerve itself located in the pons. Thus, to reach the spinal nucleus (which ● It lies in all levels of M.O, medial to the spinal tract of the trigeminal. spans the entire brain stem length) in the Caudal ● It receives pain and temperature from face, forehead. Medulla those fibers have to "descend" in what's known as the Spinal Tract of the Trigeminal ● Its tract present in all levels of M.O. is formed of descending (how its sensory and descend?see the note) fibers that terminate in the trigeminal nucleus. -

Modulation of Serotonin Signaling/Metabolism by Akkermansia Muciniphila and Its Extracellular Vesicles Through the Gut-Brain
www.nature.com/scientificreports OPEN Modulation of serotonin signaling/ metabolism by Akkermansia muciniphila and its extracellular vesicles through the gut‑brain axis in mice Rezvan Yaghoubfar1,2, Ava Behrouzi1,2, Fatemeh Ashrafan1,2, Arefeh Shahryari1,2, Hamid Reza Moradi3, Samira Choopani4, Shima Hadifar1,2, Farzam Vaziri1,2, Seyed Ali Nojoumi1,2, Abolfazl Fateh1,2*, Shohreh Khatami 5* & Seyed Davar Siadat1,2 Several studies have reported that the host‑microbe interactions in the gut modulate the host serotonin or 5‑hydroxytryptamine (5‑HT) system. Here, we evaluated the efects of Akkermansia muciniphila and its extracellular vesicles (EVs) on genes pertaining to the serotonergic system in the colon and hippocampus of mice. Male C57BL/6J mice were administered viable A. muciniphila and its EVs for 4 weeks. The serotonin levels in the colon, hippocampus, and serum of mice, as well as the human colon carcinoma cells (Caco‑2), were measured by ELISA assays. Also, the efects of A. muciniphila and its EVs on the expression of serotonin system genes in the colon and hippocampus were examined. A. muciniphila and its EVs may have a biological efect on the induction of serotonin levels in the colon and hippocampus of mice. Also, EVs increased the serotonin level in the Caco‑2 cell line. In contrast, both treatments decreased the serotonin level in the serum. Both the bacterium and its EVs had signifcant efects on the mRNA expression of genes, involved in serotonin signaling/ metabolism in the colon and hippocampus of mice. Moreover, A. muciniphila and its EVs afected the mRNA expression of infammatory cytokines (Il-10 and Tnf‑α) in the colon, however, there is no signifcant diference in infammatory cell infltrate in the histopathology of the colon. -

Serotonin Pathway in Cancer
International Journal of Molecular Sciences Review Serotonin Pathway in Cancer Pragathi Balakrishna 1, Sagila George 1, Hassan Hatoum 1,* and Sarbajit Mukherjee 2,* 1 Stephenson Cancer Center, University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA; [email protected] (P.B.); [email protected] (S.G.) 2 Roswell Park Comprehensive Cancer Center, Buffalo, NY 14263, USA * Correspondence: [email protected] (H.H.); [email protected] (S.M.) Abstract: Serotonin (5-hydroxytryptamine, 5-HT) is a biogenic monoamine produced from the essen- tial amino acid tryptophan. Serotonin’s role as a neurotransmitter in the central nervous system and a motility mediator in the gastrointestinal tract has been well defined, and its function in tumorigenesis in various cancers (gliomas, carcinoids, and carcinomas) is being studied. Many studies have shown a potential stimulatory effect of serotonin on cancer cell proliferation, invasion, dissemination, and tumor angiogenesis. Although the underlying mechanism is complex, it is proposed that serotonin levels in the tumor and its interaction with specific receptor subtypes are associated with disease progression. This review article describes serotonin’s role in cancer pathogenesis and the utility of the serotonin pathway as a potential therapeutic target in cancer treatment. Octreotide, an inhibitor of serotonin release, is used in well-differentiated neuroendocrine cancers, and the tryptophan hy- droxylase (TPH) inhibitor, telotristat, is currently being investigated in clinical trials to treat patients with metastatic neuroendocrine tumors and advanced cholangiocarcinoma. Several in vitro studies have shown the anticancer effect of 5-HT receptor antagonists in various cancers such as prostate cancer, breast cancer, urinary bladder, colorectal cancer, carcinoid, and small-cell lung cancer. -

Chapter 3: Internal Anatomy of the Central Nervous System
10353-03_CH03.qxd 8/30/07 1:12 PM Page 82 3 Internal Anatomy of the Central Nervous System LEARNING OBJECTIVES Nuclear structures and fiber tracts related to various functional systems exist side by side at each level of the After studying this chapter, students should be able to: nervous system. Because disease processes in the brain • Identify the shapes of corticospinal fibers at different rarely strike only one anatomic structure or pathway, there neuraxial levels is a tendency for a series of related and unrelated clinical symptoms to emerge after a brain injury. A thorough knowl- • Recognize the ventricular cavity at various neuroaxial edge of the internal brain structures, including their shape, levels size, location, and proximity, makes it easier to understand • Recognize major internal anatomic structures of the their functional significance. In addition, the proximity of spinal cord and describe their functions nuclear structures and fiber tracts explains multiple symp- toms that may develop from a single lesion site. • Recognize important internal anatomic structures of the medulla and explain their functions • Recognize important internal anatomic structures of the ANATOMIC ORIENTATION pons and describe their functions LANDMARKS • Identify important internal anatomic structures of the midbrain and discuss their functions Two distinct anatomic landmarks used for visual orientation to the internal anatomy of the brain are the shapes of the • Recognize important internal anatomic structures of the descending corticospinal fibers and the ventricular cavity forebrain (diencephalon, basal ganglia, and limbic (Fig. 3-1). Both are present throughout the brain, although structures) and describe their functions their shape and size vary as one progresses caudally from the • Follow the continuation of major anatomic structures rostral forebrain (telencephalon) to the caudal brainstem. -

Mechanism of Actions of Antidepressants: Beyond the Receptors
Derlemeler/Reviews Mechanism of Actions of Antidepressants: Beyond the Receptors Mechanism of Actions of Antidepressants: Beyond the Receptors Ayflegül Y›ld›z, M.D.1, Ali Saffet Gönül, M.D.2, Lut Tamam, M.D.3 ABSTRACT: MECHANISM OF ACTIONS OF ANTIDEPRESSANTS: BEYOND THE RECEPTORS Since the discovery of first antidepressants-monoamine oxidase inhibitors- a half century passed. There are now almost two- dozen antidepressant agents that work by nine distinct pharmacological mechanisms at the receptor level. However, opposite to the divergence in their pharmacological mechanisms at the receptor level, antidepressant drugs probably stimulate similar pathways in subcellular level. These subcellular events or so called beyond receptor effects are named neuroplasticity, and the mechanism may be called as adaptation. These after-receptor processes, through their effects on synaptic transmission, and gene expression are indeed capable of altering many molecular events in the brain. In this article, the mechanisms of actions of antidepressants at- and beyond- the receptors are discussed by documenting some of the evidence indicating such long-term alterations. Accordingly, the well-known effects of antidepressants on the receptor level are initiating events of antidepressant drug action, which enhance and prolong the actions of norepinephrine and/or serotonin and/or dopamine. Only if an adequate dose of an antidepressant is taken chronically, the increase in the synaptic norepinephrine and/or serotonin and/or dopamine stresses or perturbs the nervous