Genomic Signatures Reveal DNA Damage Response Deficiency In
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

LIG1 Antibody Cat
LIG1 Antibody Cat. No.: 26-847 LIG1 Antibody Specifications HOST SPECIES: Rabbit SPECIES REACTIVITY: Human, Mouse Antibody produced in rabbits immunized with a synthetic peptide corresponding a region IMMUNOGEN: of human LIG1. TESTED APPLICATIONS: ELISA, WB LIG1 antibody can be used for detection of LIG1 by ELISA at 1:62500. LIG1 antibody can be APPLICATIONS: used for detection of LIG1 by western blot at 1 μg/mL, and HRP conjugated secondary antibody should be diluted 1:50,000 - 100,000. POSITIVE CONTROL: 1) 721_B Cell Lysate PREDICTED MOLECULAR 102 kDa WEIGHT: Properties PURIFICATION: Antibody is purified by peptide affinity chromatography method. CLONALITY: Polyclonal CONJUGATE: Unconjugated PHYSICAL STATE: Liquid September 29, 2021 1 https://www.prosci-inc.com/lig1-antibody-26-847.html Purified antibody supplied in 1x PBS buffer with 0.09% (w/v) sodium azide and 2% BUFFER: sucrose. CONCENTRATION: batch dependent For short periods of storage (days) store at 4˚C. For longer periods of storage, store LIG1 STORAGE CONDITIONS: antibody at -20˚C. As with any antibody avoid repeat freeze-thaw cycles. Additional Info OFFICIAL SYMBOL: LIG1 ALTERNATE NAMES: LIG1, MGC117397, MGC130025, ACCESSION NO.: NP_000225 PROTEIN GI NO.: 4557719 GENE ID: 3978 USER NOTE: Optimal dilutions for each application to be determined by the researcher. Background and References LIG1is DNA ligase I, with functions in DNA replication and the base excision repair process. Mutations in LIG1 that lead to DNA ligase I deficiency result in immunodeficiency and increased sensitivity to DNA-damaging agents.LIG1 encodes DNA ligase I, with functions in DNA replication and the base excision repair process. -

Kinetic Analysis of Human DNA Ligase III by Justin R. Mcnally A
Kinetic Analysis of Human DNA Ligase III by Justin R. McNally A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Biological Chemistry) in the University of Michigan 2019 Doctoral Committee: Associate Professor Patrick J. O’Brien, Chair Associate Professor Bruce A. Palfey Associate Professor JoAnn M. Sekiguchi Associate Professor Raymond C. Trievel Professor Thomas E. Wilson Justin R. McNally [email protected] ORCID iD: 0000-0003-2694-2410 © Justin R. McNally 2019 Table of Contents List of Tables iii List of Figures iv Abstract vii Chapter 1 Introduction to the human DNA ligases 1 Chapter 2 Kinetic Analyses of Single-Strand Break Repair by Human DNA Ligase III Isoforms Reveal Biochemical Differences from DNA Ligase I 20 Chapter 3 The LIG3 N-terminus, in its entirety, contributes to single-strand DNA break ligation 56 Chapter 4 Comparative end-joining by human DNA ligases I and III 82 Chapter 5 A real-time DNA ligase assay suitable for high throughput screening 113 Chapter 6 Conclusions and Future Directions 137 ii List of Tables Table 2.1: Comparison of kinetic parameters for multiple turnover ligation by human DNA ligases 31 Table 2.2: Comparison of single-turnover parameters of LIG3β and LIG1 34 Table 3.1: Comparison of LIG3β N-terminal mutant kinetic parameters 67 Table 4.1: Rate constants for sequential ligation by LIG3β 95 Table 5.1: Comparison of multiple turnover kinetic parameters determined by real-time fluorescence assay and reported values 129 iii List of Figures Figure -

Evidence Supporting Dissimilatory And
University of Massachusetts Amherst ScholarWorks@UMass Amherst Microbiology Department Faculty Publication Microbiology Series 2013 Evidence supporting dissimilatory and assimilatory lignin degradation in Enterobacter lignolyticus SCF1 Kristen DeAngelis University of Massachusetts Amherst, [email protected] Deepak Sharma University of Massachusetts Amherst Rebecca Varney University of Massachusetts Amherst Blake Simmons Joint BioEnergy Institute Nancy G. Isern Environmental Molecular Sciences Laboratory See next page for additional authors Follow this and additional works at: https://scholarworks.umass.edu/micro_faculty_pubs Part of the Microbiology Commons Recommended Citation DeAngelis, Kristen; Sharma, Deepak; Varney, Rebecca; Simmons, Blake; Isern, Nancy G.; Markillie, Lye Meng; Nicora, Carrie; Norbeck, Angela D.; Taylor, Ronald C.; Aldrich, Joshua T.; and Robinson, Errol W., "Evidence supporting dissimilatory and assimilatory lignin degradation in Enterobacter lignolyticus SCF1" (2013). Frontiers in Microbiology. 303. 10.3389/fmicb.2013.00280 This Article is brought to you for free and open access by the Microbiology at ScholarWorks@UMass Amherst. It has been accepted for inclusion in Microbiology Department Faculty Publication Series by an authorized administrator of ScholarWorks@UMass Amherst. For more information, please contact [email protected]. Authors Kristen DeAngelis, Deepak Sharma, Rebecca Varney, Blake Simmons, Nancy G. Isern, Lye Meng Markillie, Carrie Nicora, Angela D. Norbeck, Ronald C. Taylor, Joshua T. Aldrich, and Errol W. Robinson This article is available at ScholarWorks@UMass Amherst: https://scholarworks.umass.edu/micro_faculty_pubs/303 ORIGINAL RESEARCH ARTICLE published: 19 September 2013 doi: 10.3389/fmicb.2013.00280 Evidence supporting dissimilatory and assimilatory lignin degradation in Enterobacter lignolyticus SCF1 Kristen M. DeAngelis 1*, Deepak Sharma 1, Rebecca Varney 1, Blake Simmons 2,3, Nancy G. -

Transcriptional Activation of Mouse Retrotransposons in Vivo: Specific Expression in Ster.Oidogenic Cells in Response to Trophlc Hormones
Downloaded from genesdev.cshlp.org on October 8, 2021 - Published by Cold Spring Harbor Laboratory Press Transcriptional activation of mouse retrotransposons in vivo: specific expression in ster.oidogenic cells in response to trophlc hormones Rachel Schiff, Ahuva Itin, and Eli Keshet Department of Virology, The Hebrew University, Hadassah Medical School, Jerusalem 91010 Israel Transcription of cellular retrotransposons is induced by a variety of physiological stimuli. We have used in situ hybridization analysis to determine the cell types in which mouse retrotransposons are transcriptionally activated in vivo under physiological conditions. Here, we report that VL30 retrotransposons are specifically expressed in steroidogenic cells within all four endocrine tissues engaged in synthesis of steroid hormones in response to the respective pituitary-derived trophic hormones. These tissues include ovarian steroidogenic theca cells and lutein cells of the corpus luteum, testosterone-producing Leydig cells of the testis, steroidogenic cells confined to the zona reticularis of the adrenal cortex, and progesterone-producing cells of the placenta. In the course of preovulatory follicular development and maturation, the profile of cells expressing the retrotransposon shifted in parallel to the changing profiles of the leutinizing hormone (LH)-induced steroidogenic output of the respective cells. Expression of VL30 in both male and female gonads was shown to be greatly stimulated by external administration of gonadotropins. In vitro studies using a LH-responsive Leydig cell line have confirmed that expression of the resident retrotransposons is gonadotropin dependent. Run-off transcription assays have indicated that activation is at the transcriptional level. To allow molecular access to gonadatropin-activated transcription units, the long terminal repeat (LTR) regulatory domains were cloned from VL30 cDNAs of LH-induced ovaries. -

3-Methyladenine DNA Glycosylases: Structure, Function, and Biological Importance Michael D
Review articles 3-Methyladenine DNA glycosylases: structure, function, and biological importance Michael D. Wyatt,1 James M. Allan,1 Albert Y. Lau,2 Tom E. Ellenberger,2 and Leona D. Samson1* Summary The genome continuously suffers damage due to its reactivity with chemical and physical agents. Finding such damage in genomes (that can be several million to several billion nucleotide base pairs in size) is a seemingly daunting task. 3-Methyladenine DNA glycosylases can initiate the base excision repair (BER) of an extraordinarily wide range of substrate bases. The advantage of such broad substrate recognition is that these enzymes provide resistance to a wide variety of DNA damaging agents; however, under certain circumstances, the eclectic nature of these enzymes can confer some biological disadvantages. Solving the X-ray crystal struc- tures of two 3-methyladenine DNA glycosylases, and creating cells and animals altered for this activity, contributes to our understanding of their enzyme mechanism and how such enzymes influence the biological response of organisms to several different types of DNA damage. BioEssays 21:668–676, 1999. 1999 John Wiley & Sons, Inc. Introduction ately interpreted by DNA-processing enzymes. Unfortunately, DNA carries life’s genetic information encoded in the arrange- these bases are also chemically reactive, and the inevitable ment of bases along the length of the DNA molecule. Each base modifications produce a variety of biological outcomes, DNA base has a distinct chemical structure that is appropri- depending on how a cell recognizes and responds to the modification. Cellular DNA repair mechanisms target these inappropriate DNA structures, and play a vital role in maintain- 1Department of Cancer Cell Biology, Harvard School of Public Health, ing genomic integrity. -

RNA Ligase Structures Reveal the Basis for RNA Specificity And
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector RNA Ligase Structures Reveal the Basis for RNA Specificity and Conformational Changes that Drive Ligation Forward Jayakrishnan Nandakumar,1,2,3 Stewart Shuman,2 and Christopher D. Lima1,* 1 Structural Biology Program 2 Molecular Biology Program 3 Training Program in Chemical Biology Sloan-Kettering Institute, New York, NY 10021, USA *Contact: [email protected] DOI 10.1016/j.cell.2006.08.038 0 SUMMARY 5 PO4 strand is DNA or RNA (Nandakumar and Shuman, 2004). Thus, ligase specificity might be dictated by dis- T4 RNA ligase 2 (Rnl2) and kinetoplastid RNA crimination of A form and B form secondary structures in 0 0 editing ligases exemplify a family of RNA repair duplex nucleic acid on either the 3 OH or 5 PO4 sides of 0 0 enzymes that seal 3 OH/5 PO4 nicks in duplex a nick. This model is difficult to extend to RNA ligases RNAs via ligase adenylylation (step 1), AMP that join single-stranded polynucleotide ends. 0 Two families of RNA ligases, named Rnl1 and Rnl2, transfer to the nick 5 PO4 (step 2), and attack by the nick 30OH on the 50-adenylylated strand function in the repair of ‘‘purposeful’’ RNA breaks. The Rnl1 family includes bacteriophage T4 RNA ligase 1 (Silber to form a phosphodiester (step 3). Crystal struc- et al., 1972; Uhlenbeck and Gumport, 1982; Wang et al., tures are reported for Rnl2 at discrete steps 2003) and tRNA ligases of fungi and plants (Wang and along this pathway: the covalent Rnl2-AMP Shuman, 2005; Englert and Beier, 2005). -

Understanding Nucleotide Excision Repair and Its Roles in Cancer and Ageing
REVIEWS DNA DAMAGE Understanding nucleotide excision repair and its roles in cancer and ageing Jurgen A. Marteijn*, Hannes Lans*, Wim Vermeulen, Jan H. J. Hoeijmakers Abstract | Nucleotide excision repair (NER) eliminates various structurally unrelated DNA lesions by a multiwise ‘cut and patch’-type reaction. The global genome NER (GG‑NER) subpathway prevents mutagenesis by probing the genome for helix-distorting lesions, whereas transcription-coupled NER (TC‑NER) removes transcription-blocking lesions to permit unperturbed gene expression, thereby preventing cell death. Consequently, defects in GG‑NER result in cancer predisposition, whereas defects in TC‑NER cause a variety of diseases ranging from ultraviolet radiation‑sensitive syndrome to severe premature ageing conditions such as Cockayne syndrome. Recent studies have uncovered new aspects of DNA-damage detection by NER, how NER is regulated by extensive post-translational modifications, and the dynamic chromatin interactions that control its efficiency. Based on these findings, a mechanistic model is proposed that explains the complex genotype–phenotype correlations of transcription-coupled repair disorders. The integrity of DNA is constantly threatened by endo of an intricate DNA-damage response (DDR), which genously formed metabolic products and by-products, comprises sophisticated repair and damage signalling such as reactive oxygen species (ROS) and alkylating processes. The DDR involves DNA-damage sensors and agents, and by its intrinsic chemical instability (for exam signalling kinases that regulate a range of downstream ple, by its ability to spontaneously undergo hydrolytic mediator and effector molecules that control repair, cell deamination and depurination). Environmental chemi cycle progression and cell fate4. The core of this DDR is cals and radiation also affect the physical constitution of formed by a network of complementary DNA repair sys DNA1. -

Mechanism of Hepatitis B Virus Cccdna Formation
viruses Review Mechanism of Hepatitis B Virus cccDNA Formation Lei Wei and Alexander Ploss * 110 Lewis Thomas Laboratory, Department of Molecular Biology, Princeton University, Washington Road, Princeton, NJ 08544, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-609-258-7128 Abstract: Hepatitis B virus (HBV) remains a major medical problem affecting at least 257 million chronically infected patients who are at risk of developing serious, frequently fatal liver diseases. HBV is a small, partially double-stranded DNA virus that goes through an intricate replication cycle in its native cellular environment: human hepatocytes. A critical step in the viral life-cycle is the conversion of relaxed circular DNA (rcDNA) into covalently closed circular DNA (cccDNA), the latter being the major template for HBV gene transcription. For this conversion, HBV relies on multiple host factors, as enzymes capable of catalyzing the relevant reactions are not encoded in the viral genome. Combinations of genetic and biochemical approaches have produced findings that provide a more holistic picture of the complex mechanism of HBV cccDNA formation. Here, we review some of these studies that have helped to provide a comprehensive picture of rcDNA to cccDNA conversion. Mechanistic insights into this critical step for HBV persistence hold the key for devising new therapies that will lead not only to viral suppression but to a cure. Keywords: hepatitis B virus; HBV; viral replication; cccDNA biogenesis; rcDNA; DNA repair Citation: Wei, L.; Ploss, A. 1. Overview of HBV Life Cycle and cccDNA Biogenesis Mechanism of Hepatitis B Virus cccDNA Formation. Viruses 2021, 13, The hepatotropic HBV belongs to the Hepadnaviridae family and is a blood-borne 1463. -

Apoptosis Signal-Regulating Kinase 1 Promotes Ochratoxin A-Induced
OPEN Apoptosis Signal-regulating Kinase 1 SUBJECT AREAS: promotes Ochratoxin A-induced renal CELL BIOLOGY RISK FACTORS cytotoxicity Rui Liang1, Xiao Li Shen1,2, Boyang Zhang1, Yuzhe Li1, Wentao Xu1, Changhui Zhao3, YunBo Luo1 Received & Kunlun Huang1 10 November 2014 Accepted 1Laboratory of food safety and molecular biology, College of Food Science and Nutritional Engineering, China Agricultural 5 January 2015 University, Beijing 100083, P.R. China, 2School of Public Health, Zunyi Medical University, Zunyi, Guizhou 563003, P.R. China, 3Department of Nutrition and Food Science, University of Maryland, College Park, MD 20742, USA. Published 28 January 2015 Oxidative stress and apoptosis are involved in Ochratoxin A (OTA)-induced renal cytotoxicity. Apoptosis signal-regulating kinase 1 (ASK1) is a Mitogen-Activated Protein Kinase Kinase Kinase (MAPKKK, Correspondence and MAP3K) family member that plays an important role in oxidative stress-induced cell apoptosis. In this study, we performed RNA interference of ASK1 in HEK293 cells and employed an iTRAQ-based requests for materials quantitative proteomics approach to globally investigate the regulatory mechanism of ASK1 in should be addressed to OTA-induced renal cytotoxicity. Our results showed that ASK1 knockdown alleviated OTA-induced ROS W.X. (xuwentao@cau. generation and Dym loss and thus desensitized the cells to OTA-induced apoptosis. We identified 33 and 24 edu.cn) differentially expressed proteins upon OTA treatment in scrambled and ASK1 knockdown cells, respectively. Pathway classification and analysis revealed that ASK1 participated in OTA-induced inhibition of mRNA splicing, nucleotide metabolism, the cell cycle, DNA repair, and the activation of lipid metabolism. We concluded that ASK1 plays an essential role in promoting OTA-induced renal cytotoxicity. -

The Dark Side of UV-Induced DNA Lesion Repair
G C A T T A C G G C A T genes Review The Dark Side of UV-Induced DNA Lesion Repair Wojciech Strzałka 1, Piotr Zgłobicki 1, Ewa Kowalska 1, Aneta Ba˙zant 1, Dariusz Dziga 2 and Agnieszka Katarzyna Bana´s 1,* 1 Department of Plant Biotechnology, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Gronostajowa 7, 30-387 Krakow, Poland; [email protected] (W.S.); [email protected] (P.Z.); [email protected] (E.K.); [email protected] (A.B.) 2 Department of Microbiology, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Gronostajowa 7, 30-387 Krakow, Poland; [email protected] * Correspondence: [email protected]; Tel.: +48-12-664-6410 Received: 27 October 2020; Accepted: 29 November 2020; Published: 2 December 2020 Abstract: In their life cycle, plants are exposed to various unfavorable environmental factors including ultraviolet (UV) radiation emitted by the Sun. UV-A and UV-B, which are partially absorbed by the ozone layer, reach the surface of the Earth causing harmful effects among the others on plant genetic material. The energy of UV light is sufficient to induce mutations in DNA. Some examples of DNA damage induced by UV are pyrimidine dimers, oxidized nucleotides as well as single and double-strand breaks. When exposed to light, plants can repair major UV-induced DNA lesions, i.e., pyrimidine dimers using photoreactivation. However, this highly efficient light-dependent DNA repair system is ineffective in dim light or at night. Moreover, it is helpless when it comes to the repair of DNA lesions other than pyrimidine dimers. -
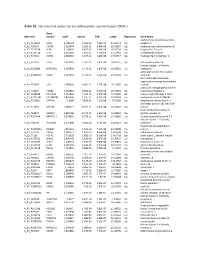
Table S1. the Statistical Metrics for Key Differentially Expressed Genes (Degs)
Table S1. The statistical metrics for key differentially expressed genes (DEGs) Gene Agilent Id Symbol logFC pValue FDR tvalue Regulation Gene Name oxidized low density lipoprotein A_24_P124624 OLR1 2.458429 1.19E-13 7.25E-10 24.04241 Up receptor 1 A_23_P90273 CHST8 2.622464 3.85E-12 6.96E-09 19.05867 Up carbohydrate sulfotransferase 8 A_23_P217528 KLF8 2.109007 4.85E-12 7.64E-09 18.76234 Up Kruppel like factor 8 A_23_P114740 CFH 2.651636 1.85E-11 1.79E-08 17.13652 Up complement factor H A_23_P34031 XAGE2 2.000935 2.04E-11 1.81E-08 17.02457 Up X antigen family member 2 A_23_P27332 TCF4 1.613097 2.32E-11 1.87E-08 16.87275 Up transcription factor 4 histone cluster 1 H1 family A_23_P250385 HIST1H1B 2.298658 2.47E-11 1.87E-08 16.80362 Up member b abnormal spindle microtubule A_33_P3288159 ASPM 2.162032 2.79E-11 2.01E-08 16.66292 Up assembly H19, imprinted maternally expressed transcript (non-protein A_24_P52697 H19 1.499364 4.09E-11 2.76E-08 16.23387 Up coding) potassium voltage-gated channel A_24_P31627 KCNB1 2.289689 6.65E-11 3.97E-08 15.70253 Up subfamily B member 1 A_23_P214168 COL12A1 2.155835 7.59E-11 4.15E-08 15.56005 Up collagen type XII alpha 1 chain A_33_P3271341 LOC388282 2.859496 7.61E-11 4.15E-08 15.55704 Up uncharacterized LOC388282 A_32_P150891 DIAPH3 2.2068 7.83E-11 4.22E-08 15.5268 Up diaphanous related formin 3 zinc finger protein 185 with LIM A_23_P11025 ZNF185 1.385721 8.74E-11 4.59E-08 15.41041 Up domain heat shock protein family B A_23_P96872 HSPB11 1.887166 8.94E-11 4.64E-08 15.38599 Up (small) member 11 A_23_P107454 -

Ligase 1 Is a Predictor of Platinum Resistance and Its Blockade Is Synthetically Lethal In
Ligase 1 is a predictor of platinum resistance and its blockade is synthetically lethal in XRCC1 deficient epithelial ovarian cancers Reem Ali1*, Muslim Alabdullah1,2*, Mashael Algethami1**, Adel Alblihy1,3**, Islam Miligy2, Ahmed Shoqafi1, Katia A. Mesquita1, Tarek Abdel-Fatah4, Stephen YT Chan4, Pei Wen Chiang5, Nigel P Mongan6,7, Emad A Rakha2, Alan E Tomkinson8, Srinivasan Madhusudan1,3*** 1 Nottingham Biodiscovery Institute, School of Medicine, University of Nottingham, University Park, Nottingham NG7 3RD, UK. 2 Department of Pathology, Division of Cancer and Stem Cells, School of Medicine, University of Nottingham, Nottingham NG51PB, UK. 3 Medical Center, King Fahad Security College (KFSC), Riyadh 11461, Saudi Arabia. 4 Department of Oncology, Nottingham University Hospitals, City Hospital Campus, Nottingham NG5 1PB, UK. 5 Department of Obstetrics & Gynaecology, Queens Medical Centre, Nottingham University Hospitals, Nottingham NG7 2UH, UK. 6 Faculty of Medicine and Health Sciences, Centre for Cancer Sciences, University of Nottingham, Sutton Bonington Campus, Sutton Bonington, Leicestershire LE12 5RD, UK 7 Department of Pharmacology, Weill Cornell Medicine, New York, NY, 10065, USA 8 Department of Internal Medicine, Division of Molecular Medicine, Health Sciences Center, The University of New Mexico, Albuquerque, NM 87102, USA. * = Joint first authors, ** = Joint second authors Running title: LIG1 targeting in ovarian cancers Declarations of interest: none Word count: 3459 Figure: 5 Table:1 *** Corresponding author: Professor Srinivasan