Transcriptome Profiles in Peripheral White Blood Cells at the Time of Artificial Insemination Discriminate Beef Heifers with Different Fertility Potential Sarah E
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Seq2pathway Vignette
seq2pathway Vignette Bin Wang, Xinan Holly Yang, Arjun Kinstlick May 19, 2021 Contents 1 Abstract 1 2 Package Installation 2 3 runseq2pathway 2 4 Two main functions 3 4.1 seq2gene . .3 4.1.1 seq2gene flowchart . .3 4.1.2 runseq2gene inputs/parameters . .5 4.1.3 runseq2gene outputs . .8 4.2 gene2pathway . 10 4.2.1 gene2pathway flowchart . 11 4.2.2 gene2pathway test inputs/parameters . 11 4.2.3 gene2pathway test outputs . 12 5 Examples 13 5.1 ChIP-seq data analysis . 13 5.1.1 Map ChIP-seq enriched peaks to genes using runseq2gene .................... 13 5.1.2 Discover enriched GO terms using gene2pathway_test with gene scores . 15 5.1.3 Discover enriched GO terms using Fisher's Exact test without gene scores . 17 5.1.4 Add description for genes . 20 5.2 RNA-seq data analysis . 20 6 R environment session 23 1 Abstract Seq2pathway is a novel computational tool to analyze functional gene-sets (including signaling pathways) using variable next-generation sequencing data[1]. Integral to this tool are the \seq2gene" and \gene2pathway" components in series that infer a quantitative pathway-level profile for each sample. The seq2gene function assigns phenotype-associated significance of genomic regions to gene-level scores, where the significance could be p-values of SNPs or point mutations, protein-binding affinity, or transcriptional expression level. The seq2gene function has the feasibility to assign non-exon regions to a range of neighboring genes besides the nearest one, thus facilitating the study of functional non-coding elements[2]. Then the gene2pathway summarizes gene-level measurements to pathway-level scores, comparing the quantity of significance for gene members within a pathway with those outside a pathway. -

Dual Proteome-Scale Networks Reveal Cell-Specific Remodeling of the Human Interactome
bioRxiv preprint doi: https://doi.org/10.1101/2020.01.19.905109; this version posted January 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Dual Proteome-scale Networks Reveal Cell-specific Remodeling of the Human Interactome Edward L. Huttlin1*, Raphael J. Bruckner1,3, Jose Navarrete-Perea1, Joe R. Cannon1,4, Kurt Baltier1,5, Fana Gebreab1, Melanie P. Gygi1, Alexandra Thornock1, Gabriela Zarraga1,6, Stanley Tam1,7, John Szpyt1, Alexandra Panov1, Hannah Parzen1,8, Sipei Fu1, Arvene Golbazi1, Eila Maenpaa1, Keegan Stricker1, Sanjukta Guha Thakurta1, Ramin Rad1, Joshua Pan2, David P. Nusinow1, Joao A. Paulo1, Devin K. Schweppe1, Laura Pontano Vaites1, J. Wade Harper1*, Steven P. Gygi1*# 1Department of Cell Biology, Harvard Medical School, Boston, MA, 02115, USA. 2Broad Institute, Cambridge, MA, 02142, USA. 3Present address: ICCB-Longwood Screening Facility, Harvard Medical School, Boston, MA, 02115, USA. 4Present address: Merck, West Point, PA, 19486, USA. 5Present address: IQ Proteomics, Cambridge, MA, 02139, USA. 6Present address: Vor Biopharma, Cambridge, MA, 02142, USA. 7Present address: Rubius Therapeutics, Cambridge, MA, 02139, USA. 8Present address: RPS North America, South Kingstown, RI, 02879, USA. *Correspondence: [email protected] (E.L.H.), [email protected] (J.W.H.), [email protected] (S.P.G.) #Lead Contact: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2020.01.19.905109; this version posted January 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Molecular Effects of Isoflavone Supplementation Human Intervention Studies and Quantitative Models for Risk Assessment
Molecular effects of isoflavone supplementation Human intervention studies and quantitative models for risk assessment Vera van der Velpen Thesis committee Promotors Prof. Dr Pieter van ‘t Veer Professor of Nutritional Epidemiology Wageningen University Prof. Dr Evert G. Schouten Emeritus Professor of Epidemiology and Prevention Wageningen University Co-promotors Dr Anouk Geelen Assistant professor, Division of Human Nutrition Wageningen University Dr Lydia A. Afman Assistant professor, Division of Human Nutrition Wageningen University Other members Prof. Dr Jaap Keijer, Wageningen University Dr Hubert P.J.M. Noteborn, Netherlands Food en Consumer Product Safety Authority Prof. Dr Yvonne T. van der Schouw, UMC Utrecht Dr Wendy L. Hall, King’s College London This research was conducted under the auspices of the Graduate School VLAG (Advanced studies in Food Technology, Agrobiotechnology, Nutrition and Health Sciences). Molecular effects of isoflavone supplementation Human intervention studies and quantitative models for risk assessment Vera van der Velpen Thesis submitted in fulfilment of the requirements for the degree of doctor at Wageningen University by the authority of the Rector Magnificus Prof. Dr M.J. Kropff, in the presence of the Thesis Committee appointed by the Academic Board to be defended in public on Friday 20 June 2014 at 13.30 p.m. in the Aula. Vera van der Velpen Molecular effects of isoflavone supplementation: Human intervention studies and quantitative models for risk assessment 154 pages PhD thesis, Wageningen University, Wageningen, NL (2014) With references, with summaries in Dutch and English ISBN: 978-94-6173-952-0 ABSTRact Background: Risk assessment can potentially be improved by closely linked experiments in the disciplines of epidemiology and toxicology. -

Genome-Wide Association Study of Diabetic Kidney Disease Highlights Biology Involved in Glomerular Basement Membrane Collagen
CLINICAL RESEARCH www.jasn.org Genome-Wide Association Study of Diabetic Kidney Disease Highlights Biology Involved in Glomerular Basement Membrane Collagen Rany M. Salem ,1 Jennifer N. Todd,2,3,4 Niina Sandholm ,5,6,7 Joanne B. Cole ,2,3,4 Wei-Min Chen,8 Darrell Andrews,9 Marcus G. Pezzolesi,10 Paul M. McKeigue,11 Linda T. Hiraki,12 Chengxiang Qiu,13 Viji Nair,14 Chen Di Liao,12 Jing Jing Cao,12 Erkka Valo ,5,6,7 Suna Onengut-Gumuscu,8 Adam M. Smiles,15 Stuart J. McGurnaghan,16 Jani K. Haukka,5,6,7 Valma Harjutsalo,5,6,7,17 Eoin P. Brennan,9 Natalie van Zuydam,18,19 Emma Ahlqvist,20 Ross Doyle,9 Tarunveer S. Ahluwalia ,21 Maria Lajer,21 Maria F. Hughes,9 Jihwan Park,13 Jan Skupien,15 Athina Spiliopoulou,11 Andrew Liu,22 Rajasree Menon,14,23 Carine M. Boustany-Kari,24 Hyun M. Kang,23,25 Robert G. Nelson,26 Ronald Klein,27 Barbara E. Klein,27 Kristine E. Lee ,27 Xiaoyu Gao,28 Michael Mauer,29 Silvia Maestroni,30 Maria Luiza Caramori,29 Ian H. de Boer ,31 Rachel G. Miller,32 Jingchuan Guo ,32 Andrew P. Boright,12 David Tregouet,33,34 Beata Gyorgy,33,34 Janet K. Snell-Bergeon,35 David M. Maahs,36 Shelley B. Bull ,37 Angelo J. Canty,38 Colin N.A. Palmer,39 Lars Stechemesser,40 Bernhard Paulweber,40 Raimund Weitgasser,40,41 Jelizaveta Sokolovska,42 Vita Rovıte,43 Valdis Pırags, 42,44 Edita Prakapiene,45 Lina Radzeviciene,46 Rasa Verkauskiene,46 Nicolae Mircea Panduru,6,47 Leif C. -

A Framework to Identify Contributing Genes in Patients with Phelan
A framework to identify contributing genes in patients with Phelan-McDermid syndrome Anne-Claude Tabet, Thomas Rolland, Marie Ducloy, Jonathan Levy, Julien Buratti, Alexandre Mathieu, Damien Haye, Laurence Perrin, Céline Dupont, Sandrine Passemard, et al. To cite this version: Anne-Claude Tabet, Thomas Rolland, Marie Ducloy, Jonathan Levy, Julien Buratti, et al.. A frame- work to identify contributing genes in patients with Phelan-McDermid syndrome. Genomic Medicine, Springer Verlag, 2017, 2, pp.32. 10.1038/s41525-017-0035-2. hal-01738521 HAL Id: hal-01738521 https://hal.umontpellier.fr/hal-01738521 Submitted on 20 Mar 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License www.nature.com/npjgenmed ARTICLE OPEN A framework to identify contributing genes in patients with Phelan-McDermid syndrome Anne-Claude Tabet 1,2,3,4, Thomas Rolland2,3,4, Marie Ducloy2,3,4, Jonathan Lévy1, Julien Buratti2,3,4, Alexandre Mathieu2,3,4, Damien Haye1, Laurence Perrin1, Céline Dupont 1, Sandrine -

Genome-Wide Association and Trans-Ethnic Meta-Analysis for Advanced Diabetic Kidney Disease: Family Investigation of Nephropathy and Diabetes (FIND)
RESEARCH ARTICLE Genome-Wide Association and Trans-ethnic Meta-Analysis for Advanced Diabetic Kidney Disease: Family Investigation of Nephropathy and Diabetes (FIND) Sudha K. Iyengar1☯*, John R. Sedor2,3☯*, Barry I. Freedman4☯*, W. H. Linda Kao5†, Matthias Kretzler6, Benjamin J. Keller6, Hanna E. Abboud7†, Sharon G. Adler8, Lyle G. Best9, Donald W. Bowden10, Allison Burlock6, Yii-Der Ida Chen11, Shelley A. Cole12, Mary E. Comeau13, Jeffrey M. Curtis14, Jasmin Divers13, Christiane Drechsler15, Ravi Duggirala12, Robert C. Elston1, Xiuqing Guo11, Huateng Huang16, Michael Marcus Hoffmann17, Barbara V. Howard18, Eli Ipp19, Paul L. Kimmel20, Michael J. Klag21, William C. Knowler14, Orly F. Kohn22, Tennille S. Leak6, David J. Leehey23, Man Li24, Alka Malhotra14, Winfried März25, Viji Nair6, Robert G. Nelson14, Susanne B. Nicholas26, Stephen J. O’Brien27, Madeleine V. Pahl28, Rulan S. Parekh29, Marcus G. Pezzolesi30, Rebekah S. Rasooly31, Charles N. Rotimi32, Jerome I. Rotter11, Jeffrey R. Schelling2, Michael F. Seldin33, Vallabh O. Shah34, Adam M. Smiles35, Michael W. Smith36, Kent D. Taylor11, Farook Thameem37¤, Denyse P. Thornley-Brown38, Barbara J. Truitt1, OPEN ACCESS Christoph Wanner39, E. Jennifer Weil14, Cheryl A. Winkler40, Philip G. Zager41, Robert P. Igo, Jr1‡, Robert L. Hanson14‡, Carl D. Langefeld11‡, Family Investigation of Citation: Iyengar SK, Sedor JR, Freedman BI, Kao Nephropathy and Diabetes (FIND)¶ WHL, Kretzler M, Keller BJ, et al. (2015) Genome- Wide Association and Trans-ethnic Meta-Analysis for 1 Department of Epidemiology & Biostatistics, Case Western Reserve University, Cleveland, Ohio, Advanced Diabetic Kidney Disease: Family United States of America, 2 Departments of Medicine, Case Western Reserve University, Cleveland, Ohio, Investigation of Nephropathy and Diabetes (FIND). -
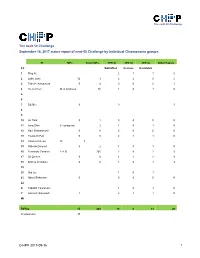
Next-MP50 Status Report
The neXt-50 Challenge The neXt-50 Challenge September 16, 2017 status report of next-50 Challenge by individual Chromosome groups. PI MPs Silver MPs JPR SI JPR SI JPR SI Other Papers Ch Submitted In press In revision 1 Ping Xu 2 1 1 0 2 Lydie Lane 12 3 2 2 0 2 3 Takeshi Kawamura 0 4 0 0 0 1 4 Yu-Ju Chen 26 in progress 86 1 0 1 0 5 6 7 Ed Nice 0 0 1 8 9 10 Jin Park 0 1 0 0 0 0 11 Jong Shin 2 in progress 2 1 0 1 0 12 Ravi Sirdeshmukh 0 0 0 0 0 0 13 Young-Ki Paik 0 5 2 1 1 0 14 CharLes Pineau 12 3 15 GiLberto Domont 0 2 1 0 1 0 16 Fernando CorraLes 2 (+ 9) 165 2 0 2 5 17 GiL Omenn 0 0 3 1 2 8 18 Andrey Archakov 0 0 1 0 1 3 19 20 Siqi Liu 1 0 1 21 Albert Sickmann 0 0 0 0 0 22 X Tadashi Yamamoto 1 0 1 0 Y Hosseini SaLekdeh 1 2 1 1 0 Mt TOTAL 15 268 19 6 13 20 (in progress) 37 C-HPP 2017-09-16 1 The neXt-50 Challenge Chromosome 1 (Ping Xu) PIC Leaders: Ping Xu, Fuchu He Contributing labs: Ping Xu, Beijing Proteome Research Center Fuchu He, Beijing Proteome Research Center Dong Yang, Beijing Proteome Research Center Wantao Ying, Beijing Proteome Research Center Pengyuan Yang, Fudan University Siqi Liu, Beijing Genome Institute Qinyu He, Jinan University Major lab members or partners contributing to the neXt50: Yao Zhang (Beijing Proteome Research Center), Yihao Wang (Beijing Proteome Research Center), Cuitong He (Beijing Proteome Research Center), Wei Wei (Beijing Proteome Research Center), Yanchang Li (Beijing Proteome Research Center), Feng Xu (Beijing Proteome Research Center), Xuehui Peng (Beijing Proteome Research Center). -

Integrated Analysis of Gene Expression Changes Associated
Miao et al. Lipids in Health and Disease (2019) 18:92 https://doi.org/10.1186/s12944-019-1032-5 RESEARCH Open Access Integrated analysis of gene expression changes associated with coronary artery disease Liu Miao1 , Rui-Xing Yin1,2,3* , Feng Huang1,2,3, Shuo Yang1, Wu-Xian Chen1 and Jin-Zhen Wu1 Abstract Background: This study investigated the pathways and genes involved in coronary artery disease (CAD) and the associated mechanisms. Methods: Two array data sets of GSE19339 and GSE56885 were downloaded. The limma package was used to analyze the differentially expressed genes (DEGs) in normal and CAD specimens. Examination of DEGs through Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment and Gene Ontology annotation was achieved by Database for Annotation, Visualization and Integrated Discovery (DAVID). The Cytoscape software facilitated the establishment of the protein-protein interaction (PPI) network and Molecular Complex Detection (MCODE) was performed for the significant modules. Results: We identified 413 DEGs (291 up-regulated and 122 down-regulated). Approximately 256 biological processes, only 1 cellular component, and 21 molecular functions were identified by GO analysis and 10 pathways were enriched by KEGG. Moreover, 264 protein pairs and 64 nodes were visualized by the PPI network. After the MCODE analysis, the top 4 high degree genes, including interleukin 1 beta (IL1B, degree = 29), intercellular adhesion molecule 1 (ICAM1, degree = 25), Jun proto-oncogene (JUN, degree = 23) and C-C motif chemokine ligand 2 (CCL2, degree = 20) had been identified to validate in RT-PCR and Cox proportional hazards regression between CAD and normals. Conclusions: The relative expression of IL1B, ICAM1 and CCL2 was higher in CAD than in normal controls (P <0.05–0. -

Role of PDZ-Binding Motif from West Nile Virus NS5 Protein on Viral
www.nature.com/scientificreports OPEN Role of PDZ‑binding motif from West Nile virus NS5 protein on viral replication Emilie Giraud1*, Chloé Otero del Val2, Célia Caillet‑Saguy2, Nada Zehrouni2, Cécile Khou5, Joël Caillet4, Yves Jacob3, Nathalie Pardigon5 & Nicolas Wolf2 West Nile virus (WNV) is a Flavivirus, which can cause febrile illness in humans that may progress to encephalitis. Like any other obligate intracellular pathogens, Flaviviruses hijack cellular protein functions as a strategy for sustaining their life cycle. Many cellular proteins display globular domain known as PDZ domain that interacts with PDZ‑Binding Motifs (PBM) identifed in many viral proteins. Thus, cellular PDZ‑containing proteins are common targets during viral infection. The non‑structural protein 5 (NS5) from WNV provides both RNA cap methyltransferase and RNA polymerase activities and is involved in viral replication but its interactions with host proteins remain poorly known. In this study, we demonstrate that the C‑terminal PBM of WNV NS5 recognizes several human PDZ‑ containing proteins using both in vitro and in cellulo high‑throughput methods. Furthermore, we constructed and assayed in cell culture WNV replicons where the PBM within NS5 was mutated. Our results demonstrate that the PBM of WNV NS5 is important in WNV replication. Moreover, we show that knockdown of the PDZ‑containing proteins TJP1, PARD3, ARHGAP21 or SHANK2 results in the decrease of WNV replication in cells. Altogether, our data reveal that interactions between the PBM of NS5 and PDZ‑containing proteins afect West Nile virus replication. Arboviruses include numerous human and animal pathogens that are important global health threats responsible for arboviroses. -

UNIVERSITY of CALIFORNIA Los Angeles a Sterile Alpha Motif
UNIVERSITY OF CALIFORNIA Los Angeles A Sterile Alpha Motif Domain Network Involved in Kidney Development A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biochemistry and Molecular Biology by Catherine Nicole Leettola 2015 ABSTRACT OF THE DISSERTATION A Sterile Alpha Motif Domain Network Involved in Kidney Development by Catherine Nicole Leettola Doctor of Philosophy in Biochemistry and Molecular Biology University of California, Los Angeles, 2015 Professor James U. Bowie, Chair Cystic kidney diseases including polycystic kidney disease (PKD) and nephronophthisis (NPHP) are the most common genetic disorders leading to end-stage renal failure in humans. Animal models and human cases of PKD and NPHP have implicated the sterile alpha motif (SAM) domain containing proteins bicaudal C homolog 1 (BICC1) and ankyrin repeat and SAM- domain containing protein 6 (ANKS6) as being involved in these conditions and important for renal development. SAM domains are known protein-protein interaction domains that are capable of binding each other to form polymers and heterodimers. Using a negGFP native gel assay, we have identified the SAM domain of the previously uncharacterized protein ankyrin repeat and SAM-domain containing protein 3 (ANKS3) as a direct binding partner of the BICC1 and ANKS6 SAM domains. We found the ANKS3 SAM domain to polymerize with moderate affinity and determined the ANKS6 SAM domain can bind to a single end of this polymer. Crystal structures of the ANKS3 SAM domain polymer and the ANKS3 SAM-ANKS6 SAM ii heterodimer are presented to reveal typical ML-EH SAM domain interaction interfaces with a pronounced charge complementarity. -

Transparent Machine Learning for Multi-Omics Analysis of Mental Disorders
UPTEC X20 015 Examensarbete 30 hp Juni 2020 Transparent Machine Learning for Multi-Omics Analysis of Mental Disorders Stella Belin Abstract Transparent Machine Learning for Multi-Omics Analysis of Mental Disorders Stella Belin Teknisk- naturvetenskaplig fakultet UTH-enheten Schizophrenia and bipolar disorder are two severe mental disorders that affect more than 65 million individuals worldwide. The aim of this Besöksadress: project was to find co-prediction mechanisms for genes associated with Ångströmlaboratoriet Lägerhyddsvägen 1 schizophrenia and bipolar disorder using a multi-omics data set and a Hus 4, Plan 0 transparent machine learning approach. The overall purpose of the project was to further understand the biological mechanisms of these Postadress: complex disorders. In this work, publicly available multi-omics data Box 536 751 21 Uppsala collected from post-mortem brain tissue were used. The omics types included were gene expression, DNA methylation, and SNP array data. The Telefon: data consisted of samples from individuals with schizophrenia, bipolar 018 – 471 30 03 disorder, and healthy controls. Individuals with schizophrenia or Telefax: bipolar disorder were considered as a combined CASE class. 018 – 471 30 00 Using machine learning techniques, a multi-omics pipeline was developed Hemsida: to integrate these data in a manner such that all types were adequately http://www.teknat.uu.se/student represented. A feature selection was performed on methylation and SNP data, where the most important sites were estimated and mapped to their corresponding genes. Next, those genes were intersected with the gene expression data, and another feature selection was performed on the gene expression data. The most important genes were used to develop an interpretable rule-based model with an accuracy of 88%. -

Content Based Search in Gene Expression Databases and a Meta-Analysis of Host Responses to Infection
Content Based Search in Gene Expression Databases and a Meta-analysis of Host Responses to Infection A Thesis Submitted to the Faculty of Drexel University by Francis X. Bell in partial fulfillment of the requirements for the degree of Doctor of Philosophy November 2015 c Copyright 2015 Francis X. Bell. All Rights Reserved. ii Acknowledgments I would like to acknowledge and thank my advisor, Dr. Ahmet Sacan. Without his advice, support, and patience I would not have been able to accomplish all that I have. I would also like to thank my committee members and the Biomed Faculty that have guided me. I would like to give a special thanks for the members of the bioinformatics lab, in particular the members of the Sacan lab: Rehman Qureshi, Daisy Heng Yang, April Chunyu Zhao, and Yiqian Zhou. Thank you for creating a pleasant and friendly environment in the lab. I give the members of my family my sincerest gratitude for all that they have done for me. I cannot begin to repay my parents for their sacrifices. I am eternally grateful for everything they have done. The support of my sisters and their encouragement gave me the strength to persevere to the end. iii Table of Contents LIST OF TABLES.......................................................................... vii LIST OF FIGURES ........................................................................ xiv ABSTRACT ................................................................................ xvii 1. A BRIEF INTRODUCTION TO GENE EXPRESSION............................. 1 1.1 Central Dogma of Molecular Biology........................................... 1 1.1.1 Basic Transfers .......................................................... 1 1.1.2 Uncommon Transfers ................................................... 3 1.2 Gene Expression ................................................................. 4 1.2.1 Estimating Gene Expression ............................................ 4 1.2.2 DNA Microarrays ......................................................