Signature Redacted Signature of Author
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transport of Dangerous Goods
ST/SG/AC.10/1/Rev.16 (Vol.I) Recommendations on the TRANSPORT OF DANGEROUS GOODS Model Regulations Volume I Sixteenth revised edition UNITED NATIONS New York and Geneva, 2009 NOTE The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. ST/SG/AC.10/1/Rev.16 (Vol.I) Copyright © United Nations, 2009 All rights reserved. No part of this publication may, for sales purposes, be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, electrostatic, magnetic tape, mechanical, photocopying or otherwise, without prior permission in writing from the United Nations. UNITED NATIONS Sales No. E.09.VIII.2 ISBN 978-92-1-139136-7 (complete set of two volumes) ISSN 1014-5753 Volumes I and II not to be sold separately FOREWORD The Recommendations on the Transport of Dangerous Goods are addressed to governments and to the international organizations concerned with safety in the transport of dangerous goods. The first version, prepared by the United Nations Economic and Social Council's Committee of Experts on the Transport of Dangerous Goods, was published in 1956 (ST/ECA/43-E/CN.2/170). In response to developments in technology and the changing needs of users, they have been regularly amended and updated at succeeding sessions of the Committee of Experts pursuant to Resolution 645 G (XXIII) of 26 April 1957 of the Economic and Social Council and subsequent resolutions. -

Hazardous Material Inventory Statement
City of Brooklyn Park FIRE DEPARTMENT 5200 - 85th Avenue North Brooklyn Park MN 55443 Phone: (763)493-8020 Fax: (763) 493-8391 Hazardous Materials Inventory Statement Users Guide A separate inventory statement shall be provided for each building. An amended inventory statement shall be provided within 30 days of the storage of any hazardous materials or plastics that changes or adds a hazard class or which is sufficient in quantity to cause an increase in the quantity which exceeds 5 percent for any hazard class. The hazardous materials inventory statement shall list by hazard class categories. Each grouping shall provide the following information for each hazardous material listed for that group including a total quantity for each group of hazard class. 1. Hazard class. (See attached Hazardous Materials Categories Listing) 2. Common or trade name. 3. Chemical Abstract Service Number (CAS number) found in 29 Code of Federal Regulations (C.F.R.). 4. Whether the material is pure or a mixture, and whether the material is a solid, liquid or gas 5. Maximum aggregate quantity stored at any one time. 6. Maximum aggregate quantity In-Use (Open to atmosphere) at any one time. 7. Maximum aggregate quantity In-Use (Closed to atmosphere) at any one time. 8. Storage conditions related to the storage type, high-pile, encapsulated, non-encapsulated. Attached is a listing of categories that all materials need to be organized to. Definitions of these categories are also attached for your use. At the end of this packet are blank forms for completing this project. For questions regarding Hazardous Materials Inventory Statement contact the Fire Department at 763-493-8020. -

Towards Better Understanding of C60 Organosols†
PCCP View Article Online PAPER View Journal | View Issue Towards better understanding of C60 organosols† a a a Cite this: Phys. Chem. Chem. Phys., Nikolay O. Mchedlov-Petrossyan,* Nika N. Kamneva, Younis T. M. Al-Shuuchi, b c d e 2016, 18,2517 Andriy I. Marynin, Olexii S. Zozulia, Alexander P. Kryshtal, Vladimir K. Klochkov and Sergey V. Shekhovtsova It is of common knowledge that fullerenes form colloids in polar solvents. However, the coagulation via electrolytes and the origin of the negative charge of species are still unexplored. Using a ‘radical À scavenger’ and electrospray ionization spectroscopy (ESI), we proved the formation of ion-radical C60 2À 2À and its (probable) transformation into C60 or (C60)2 . The coagulation of C60 organosols by NaClO4 and other perchlorates and nitrates in acetonitrile and its mixture with benzene obeys the Schulze– Hardy rule. At higher Ca(ClO4)2 and La(ClO4)3 concentrations, instead of coagulation, stable re-charged Received 7th November 2015, colloidal particles appeared, up to a zeta-potential of +(20–42) mV, as compared with À(33–35) mV of Accepted 7th December 2015 the initial organosols. The influence of both HClO4 and CF3SO3H was similar. This phenomenon is attrib- DOI: 10.1039/c5cp06806a uted to poor solvation of inorganic cations in cationo- and protophobic acetonitrile, which was proven using [2.2.2] cryptand. Further increasing the concentration of Ca(ClO4)2 led again to coagulation, thus www.rsc.org/pccp demonstrating a novel type of ‘coagulation zones’. 1. Introduction believe that -

Layered Iron Vanadate As a High-Capacity Cathode Material for Nonaqueous Calcium-Ion Batteries
batteries Article Layered Iron Vanadate as a High-Capacity Cathode Material for Nonaqueous Calcium-Ion Batteries Munseok S. Chae 1 , Dedy Setiawan 1, Hyojeong J. Kim 1 and Seung-Tae Hong 1,2,* 1 Department of Energy Science and Engineering, DGIST (Daegu Gyeongbuk Institute of Science and Technology), Daegu 42988, Korea; [email protected] (M.S.C.); [email protected] (D.S.); [email protected] (H.J.K.) 2 Energy Science and Engineering Research Center, DGIST (Daegu Gyeongbuk Institute of Science and Technology), Daegu 42988, Korea * Correspondence: [email protected]; Tel.: +82-53-785-6415 Abstract: Calcium-ion batteries represent a promising alternative to the current lithium-ion batteries. Nevertheless, calcium-ion intercalating materials in nonaqueous electrolytes are scarce, probably due to the difficulties in finding suitable host materials. Considering that research into calcium-ion batteries is in its infancy, discovering and characterizing new host materials would be critical to further development. Here, we demonstrate FeV3O9·1.2H2O as a high-performance calcium-ion battery cathode material that delivers a reversible discharge capacity of 303 mAh g−1 with a good cycling stability and an average discharge voltage of ~2.6 V (vs. Ca/Ca2+). The material was synthesized via a facile co-precipitation method. Its reversible capacity is the highest among calcium- ion battery materials, and it is the first example of a material with a capacity much larger than that of conventional lithium-ion battery cathode materials. Bulk intercalation of calcium into the host lattice contributed predominantly to the total capacity at a lower rate, but became comparable to that due to Citation: Chae, M.S.; Setiawan, D.; surface adsorption at a higher rate. -

Maine Remedial Action Guidelines (Rags) for Contaminated Sites
Maine Department of Environmental Protection Remedial Action Guidelines for Contaminated Sites (RAGs) Effective Date: May 1, 2021 Approved by: ___________________________ Date: April 27, 2021 David Burns, Director Bureau of Remediation & Waste Management Executive Summary MAINE DEPARTMENT OF ENVIRONMENTAL PROTECTION 17 State House Station | Augusta, Maine 04333-0017 www.maine.gov/dep Maine Department of Environmental Protection Remedial Action Guidelines for Contaminated Sites Contents 1 Disclaimer ...................................................................................................................... 1 2 Introduction and Purpose ............................................................................................... 1 2.1 Purpose ......................................................................................................................................... 1 2.2 Consistency with Superfund Risk Assessment .............................................................................. 1 2.3 When to Use RAGs and When to Develop a Site-Specific Risk Assessment ................................. 1 3 Applicability ................................................................................................................... 2 3.1 Applicable Programs & DEP Approval Process ............................................................................. 2 3.1.1 Uncontrolled Hazardous Substance Sites ............................................................................. 2 3.1.2 Voluntary Response Action Program -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -

Fedex Ground Hazardous Materials Shipping Guide Is Intended to Simplify Title 49 CFR
FedEx Ground Package Systems Inc. is committed to the safe transportation of hazardous materials. It is very important that each person engaged in the transportation of hazardous materials has the proper training and is thoroughly familiar with the Title 49CFR (Code of Federal Regulations) and/or USPS Publication 52. This guide is intended only to assist you in your preparation of hazardous materials shipped via FedEx Ground Package Systems Inc. It is the shipper’s responsibility to ensure each hazardous material package is in compliance with applicable Department of Transportation (D.O.T.) regulations and FedEx Ground Package Systems Inc. requirements. Failure to comply with these regulations and requirements may subject the shipper and carrier to fines and penalties. Improperly prepared hazmat packages or documentation may be subject to an additional charge(s) due to the unexpected hanlding associated with these shipments. Due to the changing nature of D.O.T. regulations and other information, it is impossible to guarantee absolute accuracy of the material contained in this guide. FedEx Ground Package Systems Inc., therefore, cannot assume any responsibility for omissions, errors, misprinting, or ambiguity contained within this guide and shall not be held liable in any degree for any loss or injury caused by such omission or error presented in this publication. Shippers should consult the most current version of the hazardous material regulations. Training is mandatory for those shipping hazardous materials, including limited quantity and other exceptions. The www.shipsafeshipsmart.com battery and hazmat training programs offer shippers an economical source of basic ground battery and/or hazardous materials shipping as well as addressing FedEx Ground specific issues. -

Semicon Cupla Seal Material Selection Table
Seal Material Selection Table (For reference) For seal parts in the Cupla (the important parts that prevent leaking to the outside), it is important to select the most appropriate seal material to suit the property and temperature of the fluid. It is so important that wrong selection may not only completely malfunction the Cupla but also cause an unexpected accident. *The rubber material must be selected and specified by the user. Seal Material Seal Material Seal Material Fluids Fluids Fluids Fluoro Fluoro Fluoro rubber rubber rubber rubber rubber rubber Ethylene- Ethylene- Ethylene- propylene propylene propylene Perfluoroelastomer Perfluoroelastomer Perfluoroelastomer A Acetaldehyde ─ ○ ◎ B Bromine water ◎ ─ D Dichlorophenol ◎ ─ Acetic anhydride ─ ○ ◎ Butadiene ○ △ Diethanolamine ─ ○ Acetone ─ ◎ ◎ Butane ◎ ─ Diethylene glycol ◎ ◎ Acetonitrile ─ ○ Butane (liquid) ◎ ─ E Ethanol ◎ ◎ ◎ Acetophenone ─ ◎ ◎ Butane (2.2-, 3-dimethyl) ◎ ─ Ethyl acetate ─ ○ Acetylacetone ─ ◎ ◎ Butanol (Butyl alcohol) ◎ ○ Ethyl alcohol ◎ ◎ ◎ Acetyl chloride ◎ ─ Butyl acetate ─ ○ ○ Ethyl benzene ◎ ─ ◎ Air (50˚C) ◎ ◎ ◎ Butyl stearate ◎ ─ Ethyl cellulose ─ ○ Aluminium bromide (65˚C) ◎ ◎ Butylene ◎ ─ Ethyl chloride ◎ ◎ Aluminium chloride (65˚C) ◎ ◎ Butyraldehyde ─ ○ ○ Ethylene glycol ◎ ◎ ◎ Aluminium nitrate (65˚C) ─ ◎ C Calcium acetate ─ ◎ Ethylene trichloride ◎ ─ Aluminium sulfate (65˚C) ◎ ◎ Calcium acetate (65˚C) ─ ◎ F Fluorine (dry ) ─ ─ ○ Amine ─ ○ ○ Calcium carbide ─ ─ Formaldehyde ─ ─ Ammonia (anhydrous) ─ ◎ Calcium carbonate ─ ─ Furfural ─ ○ ◎ Ammonia (cool) -

Calcium Perchlorate Hazard Summary Identification
Common Name: CALCIUM PERCHLORATE CAS Number: 13477-36-6 RTK Substance number: 0326 DOT Number: UN 1455 Date: February 2001 --------------------------------------------------------------------------- --------------------------------------------------------------------------- HAZARD SUMMARY WORKPLACE EXPOSURE LIMITS * No acute (short-term) or chronic (long-term) health effects No occupational exposure limits have been established for are known at this time. Calcium Perchlorate. This does not mean that this substance is not harmful. Safe work practices should always be IDENTIFICATION followed. Calcium Perchlorate is a white (sand-like) solid. WAYS OF REDUCING EXPOSURE REASON FOR CITATION * Where possible, enclose operations and use local exhaust * Calcium Perchlorate is on the Hazardous Substance List ventilation at the site of chemical release. If local exhaust because it is cited by DOT. ventilation or enclosure is not used, respirators should be * Definitions are provided on page 5. worn. * Wear protective work clothing. HOW TO DETERMINE IF YOU ARE BEING * Wash thoroughly immediately after exposure to Calcium EXPOSED Perchlorate. The New Jersey Right to Know Act requires most employers * Post hazard and warning information in the work area. In to label chemicals in the workplace and requires public addition, as part of an ongoing education and training employers to provide their employees with information and effort, communicate all information on the health and training concerning chemical hazards and controls. The safety hazards of Calcium Perchlorate to potentially federal OSHA Hazard Communication Standard, 1910.1200, exposed workers. requires private employers to provide similar training and information to their employees. * Exposure to hazardous substances should be routinely evaluated. This may include collecting personal and area air samples. You can obtain copies of sampling results from your employer. -

Chapter 4 Nomenclature
Chapter 4 Nomenclature 1. A binary compound is one that contains only two elements. Examples are sodium chloride, water, and carbon dioxide. 2. compounds that contain a metal and a nonmetal; compounds containing two nonmetals 3. cation, anion 4. cation 5. The substance “sodium chloride” consists of an extended lattice array of sodium ions, Na+, and chloride ions, Cl–. Each sodium ion is surrounded by several chloride ions, and each chloride ion is surrounded by several sodium ions. We write the formula as NaCl to indicate the relative number of each ion in the substance. 6. a. sodium iodide b. calcium fluoride c. aluminum sulfide d. calcium bromide e. strontium oxide f. silver chloride [silver(I) chloride] g. cesium iodide h. lithium oxide 7. a. Incorrect; BaH2 is barium hydride. b. Incorrect; Na2O is sodium oxide. c. Correct d. Incorrect; SiO2 is silicon dioxide. e. Correct 8. a. Since each iodide ion has a 1– charge, the iron ion must have a 3+ charge: the name is iron(III) iodide. b. Since each chloride ion has a 1– charge, the manganese must have a 2+ charge: the name is manganese(II) chloride. c. Since the oxide ion has a 2– charge, the mercury ion must have a 2+ charge: the name is mercury(II) oxide. d. Since the oxide ion has a 2– charge, the copper atoms must each have a 1+ charge: the name is copper(I) oxide. e. Since the oxide ion has a 2– charge, the copper ion must have a 2+ charge: copper(II) oxide. f. -
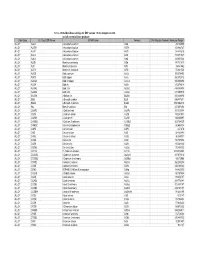
Database Full Listing
16-Nov-06 OLI Data Base Listings for ESP version 7.0.46, Analyzers 2.0.46 and all current alliance products Data Base OLI Tag (ESP) Name IUPAC Name Formula CAS Registry Number Molecular Weight ALLOY AL2U 2-Aluminum uranium Al2U 291.98999 ALLOY AL3TH 3-Aluminum thorium Al3Th 312.982727 ALLOY AL3TI 3-Aluminum titanium Al3Ti 128.824615 ALLOY AL3U 3-Aluminum uranium Al3U 318.971527 ALLOY AL4U 4-Aluminum uranium Al4U 345.953064 ALLOY ALSB Aluminum antimony AlSb 148.731537 ALLOY ALTI Aluminum titanium AlTi 74.861542 ALLOY ALTI3 Aluminum 3-titanium AlTi3 170.621536 ALLOY AUCD Gold cadmium AuCd 309.376495 ALLOY AUCU Gold copper AuCu 260.512512 ALLOY AUCU3 Gold 3-copper AuCu3 387.604492 ALLOY AUSN Gold tin AuSn 315.676514 ALLOY AUSN2 Gold 2-tin AuSn2 434.386505 ALLOY AUSN4 Gold 4-tin AuSn4 671.806519 ALLOY BA2SN 2-Barium tin Ba2Sn 393.369995 ALLOY BI2U 2-Bismuth uranium Bi2U 655.987671 ALLOY BI4U3 4-Bismuth 3-uranium Bi4U3 1550.002319 ALLOY BIU Bismuth uranium BiU 447.007294 ALLOY CA2PB 2-Calcium lead Ca2Pb 287.355988 ALLOY CA2SI 2-Calcium silicon Ca2Si 108.241501 ALLOY CA2SN 2-Calcium tin Ca2Sn 198.865997 ALLOY CA3SB2 3-Calcium 2-antimony Ca3Sb2 363.734009 ALLOY CAMG2 Calcium 2-magnesium CaMg2 88.688004 ALLOY CAPB Calcium lead CaPb 247.278 ALLOY CASI Calcium silicon CaSi 68.163498 ALLOY CASI2 Calcium 2-silicon CaSi2 96.249001 ALLOY CASN Calcium tin CaSn 158.787994 ALLOY CAZN Calcium zinc CaZn 105.468002 ALLOY CAZN2 Calcium 2-zinc CaZn2 170.858002 ALLOY CD11U 11-Cadmium uranium Cd11U 1474.536865 ALLOY CD3AS2 3-Cadmium 2-arsenic As2Cd3 487.073212 -

Magnesium Sulfate As a Key Mineral for the Detection of Organic Molecules on Mars Using Pyrolysis Pascaline François, Cyril Szopa, Arnaud Buch, Patrice Coll, Amy C
Magnesium sulfate as a key mineral for the detection of organic molecules on Mars using pyrolysis Pascaline François, Cyril Szopa, Arnaud Buch, Patrice Coll, Amy C. Mcadam, Paul R. Mahaffy, Caroline Freissinet, Daniel P. Glavin, Rafael Navarro-Gonzalez, Michel Cabane To cite this version: Pascaline François, Cyril Szopa, Arnaud Buch, Patrice Coll, Amy C. Mcadam, et al.. Magnesium sulfate as a key mineral for the detection of organic molecules on Mars using pyrolysis. Journal of Geophysical Research. Planets, Wiley-Blackwell, 2016, 121 (1), pp.61-74. 10.1002/2015JE004884. insu-01249128 HAL Id: insu-01249128 https://hal-insu.archives-ouvertes.fr/insu-01249128 Submitted on 12 Sep 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. PUBLICATIONS Journal of Geophysical Research: Planets RESEARCH ARTICLE Magnesium sulfate as a key mineral 10.1002/2015JE004884 for the detection of organic molecules Key Points: on Mars using pyrolysis • Magnesium sulfate leads to oxidation of organic molecules during pyrolysis P. François1, C. Szopa2, A. Buch3, P. Coll1, A. C. McAdam4, P. R. Mahaffy4, C. Freissinet5, D. P. Glavin4, • Trapped organics are protected from 6 2 oxidation from perchlorate R.