This Thesis Has Been Submitted in Fulfilment of the Requirements for a Postgraduate Degree (E.G
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Identification of the Binding Partners for Hspb2 and Cryab Reveals
Brigham Young University BYU ScholarsArchive Theses and Dissertations 2013-12-12 Identification of the Binding arP tners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non- Redundant Roles for Small Heat Shock Proteins Kelsey Murphey Langston Brigham Young University - Provo Follow this and additional works at: https://scholarsarchive.byu.edu/etd Part of the Microbiology Commons BYU ScholarsArchive Citation Langston, Kelsey Murphey, "Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins" (2013). Theses and Dissertations. 3822. https://scholarsarchive.byu.edu/etd/3822 This Thesis is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston A thesis submitted to the faculty of Brigham Young University in partial fulfillment of the requirements for the degree of Master of Science Julianne H. Grose, Chair William R. McCleary Brian Poole Department of Microbiology and Molecular Biology Brigham Young University December 2013 Copyright © 2013 Kelsey Langston All Rights Reserved ABSTRACT Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactors and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston Department of Microbiology and Molecular Biology, BYU Master of Science Small Heat Shock Proteins (sHSP) are molecular chaperones that play protective roles in cell survival and have been shown to possess chaperone activity. -
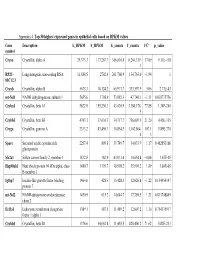
Appendix 4. Top 50 Highest Expressed Genes in Epithelial Cells Based on RPKM Values
Appendix 4. Top 50 highest expressed genes in epithelial cells based on RPKM values Gene Description E_RPKM F_RPKM E_counts F_counts FC* p_value symbol Cryaa Crystallin, alpha A 29,373.3 177,267.7 366,616.4 6,264,319. 17.09 9.11E-118 1 RP23– Long intergenic non-coding RNA 11,888.5 2702.4 261,760.9 134,763.0 −1.94 1 81C12.3 Cryab Crystallin, alpha B 5673.3 10,124.2 65,971.7 333,597.9 5.06 2.71E-43 mt-Nd1 NADH dehydrogenase, subunit 1 5655.6 1798.9 53,082.3 47,748.1 −1.11 0.838775756 Cryba1 Crystallin, beta A1 5622.0 155,230.3 43,420.9 3,380,176. 77.85 1.34E-240 5 Crybb3 Crystallin, beta B3 4743.1 37,636.3 34,717.7 736,007.9 21.20 4.45E-135 Cryga Crystallin, gamma A 2333.2 83,496.3 10,854.5 1,162,864. 107.1 5.89E-270 6 3 Sparc Secreted acidic cysteine rich 2257.4 809.8 39,749.7 34,033.9 −1.17 0.462853166 glycoprotein Slc2a1 Solute carrier family 2, member 1 1832.8 162.9 43,031.4 10,654.8 −4.04 1.67E-05 Hsp90ab1 Heat shock protein 90 kDa alpha, class 1480.7 1139.7 18,998.2 35,901.2 1.89 3.84E-05 B member 1 Igfbp7 Insulin-like growth factor binding 1464.6 428.3 15,428.3 12,626.8 −1.22 0.154954147 protein 7 mt-Nd2 NADH-ubiquinone oxidoreductase 1450.9 615.2 14,644.7 17,789.5 1.21 0.833748849 chain 2 Eef1a1 Eukaryotic translation elongation 1389.1 587.5 11,489.2 12,607.2 1.10 0.754135917 factor 1 alpha 1 Crybb1 Crystallin, beta B1 1376.6 34,662.8 11,455.5 820,406.2 71.62 5.82E-233 Htra3 HtrA serine peptidase 3 1338.6 162.0 23,197.6 6433.9 −3.61 3.93E-05 Gnb2l1 Guanine nucleotide-binding protein 1293.3 670.1 14,495.1 21,652.1 1.49 0.001685952 -

A Comprehensive Analysis of the Expression of Crystallins in Mouse Retina Jinghua Xi Washington University School of Medicine in St
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2003 A comprehensive analysis of the expression of crystallins in mouse retina Jinghua Xi Washington University School of Medicine in St. Louis Rafal Farjo University of Michigan - Ann Arbor Shigeo Yoshida University of Michigan - Ann Arbor Timothy S. Kern Case Western Reserve University Anand Swaroop University of Michigan - Ann Arbor See next page for additional authors Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Xi, Jinghua; Farjo, Rafal; Yoshida, Shigeo; Kern, Timothy S.; Swaroop, Anand; and Andley, Usha P., ,"A comprehensive analysis of the expression of crystallins in mouse retina." Molecular Vision.9,. 410-419. (2003). https://digitalcommons.wustl.edu/open_access_pubs/1801 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. Authors Jinghua Xi, Rafal Farjo, Shigeo Yoshida, Timothy S. Kern, Anand Swaroop, and Usha P. Andley This open access publication is available at Digital Commons@Becker: https://digitalcommons.wustl.edu/open_access_pubs/1801 Molecular Vision 2003; 9:410-9 <http://www.molvis.org/molvis/v9/a53> © 2003 Molecular Vision Received 28 May 2003 | Accepted 19 August 2003 | Published 28 August 2003 A comprehensive analysis of the expression of crystallins in mouse retina Jinghua Xi,1 Rafal Farjo,3 Shigeo Yoshida,3 Timothy S. Kern,5 Anand Swaroop,3,4 Usha P. Andley1,2 Departments of 1Ophthalmology and Visual Sciences and 2Biochemistry and Molecular Biophysics, Washington University School of Medicine, St. -

Related Macular Degeneration and Cutis Laxa
UvA-DARE (Digital Academic Repository) Genetic studies of age-related macular degeneration Baas, D.C. Publication date 2012 Document Version Final published version Link to publication Citation for published version (APA): Baas, D. C. (2012). Genetic studies of age-related macular degeneration. General rights It is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), other than for strictly personal, individual use, unless the work is under an open content license (like Creative Commons). Disclaimer/Complaints regulations If you believe that digital publication of certain material infringes any of your rights or (privacy) interests, please let the Library know, stating your reasons. In case of a legitimate complaint, the Library will make the material inaccessible and/or remove it from the website. Please Ask the Library: https://uba.uva.nl/en/contact, or a letter to: Library of the University of Amsterdam, Secretariat, Singel 425, 1012 WP Amsterdam, The Netherlands. You will be contacted as soon as possible. UvA-DARE is a service provided by the library of the University of Amsterdam (https://dare.uva.nl) Download date:05 Oct 2021 G������ S������ �� A��-������� M������ D����������� D����������� M������ G������ S������ �� A��-������� | 2012 D�������� C. B��� G������ S������ �� A��-������� M������ D����������� D�������� C. B��� cover.indd 1 31-10-12 08:36 Genetic Studies of Age-related Macular Degeneration Dominique C. Baas Chapter 0.indd 1 23-10-12 19:24 The research described in this thesis was conducted at the Netherlands Institute for Neuroscience (NIN), an institute of the Royal Netherlands Academy of Arts and Sciences, Department of Clinical and Molecular Ophthalmogenetics, Amsterdam, The Netherlands. -

Method for Diagnosing Non-Small Cell Lung Cancers
(19) TZZ Z _ T (11) EP 2 270 221 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 05.01.2011 Bulletin 2011/01 C12Q 1/68 (2006.01) C12N 15/12 (2006.01) G01N 33/50 (2006.01) C12N 15/11 (2006.01) (2006.01) (2006.01) (21) Application number: 10010329.0 A61P 35/00 A61K 39/00 (22) Date of filing: 22.09.2003 (84) Designated Contracting States: (72) Inventors: AT BE BG CH CY CZ DE DK EE ES FI FR GB GR • Nakamura, Yusuke HU IE IT LI LU MC NL PT RO SE SI SK TR Yokohama-shi Kanagawa 225-0011 (JP) (30) Priority: 30.09.2002 US 414673 P • Daigo, Yataro 28.02.2003 US 451374 P Yokohama-shi 28.04.2003 US 466100 P Kanagawa 222-0031 (JP) • Nakatsuru, Shuichi (62) Document number(s) of the earlier application(s) in Saitama-shi accordance with Art. 76 EPC: Saitama 338-0002 (JP) 06022167.8 / 1 743 947 03753941.8 / 1 551 998 (74) Representative: Vossius & Partner Siebertstrasse 4 (71) Applicant: Oncotherapy Science, Inc. 81675 München (DE) Kawasaki-shi Kanagawa 213-0012 (JP) Remarks: This application was filed on 23-09-2010 as a divisional application to the application mentioned under INID code 62. (54) Method for diagnosing non-small cell lung cancers (57) Disclosed are methods for detecting non-small cancerous tissues are provided. Also disclosed are meth- cell lung cancer using differentially expressed genes. ods of identifying compounds for treating and preventing Furthermore, novel human genes whose expression is non-small cell lung cancer. -

Congenital Cataracts Due to a Novel 2‑Bp Deletion in CRYBA1/A3
1614 MOLECULAR MEDICINE REPORTS 10: 1614-1618, 2014 Congenital cataracts due to a novel 2‑bp deletion in CRYBA1/A3 JING ZHANG1, YANHUA ZHANG1, FANG FANG1, WEIHONG MU1, NING ZHANG2, TONGSHUN XU3 and QINYING CAO1 1Prenatal Diagnosis Center, Shijiazhuang Obstetrics and Gynecology Hospital; 2Department of Cardiology, The Second Hospital of Hebei Medical University; 3Department of Surgery, Shijiazhuang Obstetrics and Gynecology Hospital, Shijiazhuang, Hebei, P.R. China Received September 22, 2013; Accepted April 11, 2014 DOI: 10.3892/mmr.2014.2324 Abstract. Congenital cataracts, which are a clinically and located in the eye lens. The major human crystallins comprise genetically heterogeneous group of eye disorders, lead to 90% of protein in the mature lens and contain two different visual impairment and are a significant cause of blindness superfamilies: the small heat‑shock proteins (α-crystallins) in childhood. A major proportion of the causative mutations and the βγ-crystallins. for congenital cataracts are found in crystallin genes. In the In this study a functional candidate approach was used present study, a novel deletion mutation (c.590-591delAG) in to investigate the known crystallin genes, including CRYAA, exon 6 of CRYBA1/A3 was identified in a large family with CRYAB, CRYBA1/A3, CRYBB1, CRYBB2, CRYGC, CRYGD autosomal dominant congenital cataracts. An increase in and CRYGS, in which a major proportion of the mutations local hydrophobicity was predicted around the mutation site; identified in a large family with congenital cataracts were however, further studies are required to determine the exact found. effect of the mutation on βA1/A3-crystallin structure and function. To the best of our knowledge, this is the first report Subjects and methods of an association between a frameshift mutation in exon 6 of CRYBA1/A3 and congenital cataracts. -

Congenital Eye Disorders Gene Panel
Congenital eye disorders gene panel Contact details Introduction Regional Genetics Service Ocular conditions are highly heterogeneous and show considerable phenotypic overlap. 1 in Levels 4-6, Barclay House 2,500 children in the UK are diagnosed as blind or severely visually impaired by the time they 37 Queen Square reach one year old. As many as half of these cases are likely to be inherited and remain undiagnosed due to the vast number of genes involved in these conditions. Many congenital London, WC1N 3BH eye disorders causing visual impairment or blindness at birth or progressive visual impairment T +44 (0) 20 7762 6888 also include syndromic conditions involving additional metabolic, developmental, physical or F +44 (0) 20 7813 8578 sensory abnormalities. Gene panels offer the enhanced probability of diagnosis as a very large number of genes can be interrogated. Samples required Ocular birth defects include all inheritance modalities. Autosomal dominant and recessive 5ml venous blood in plastic EDTA diseases as well as X-linked dominant and recessive diseases are seen. These conditions can bottles (>1ml from neonates) also be caused by de novo variants. Prenatal testing must be arranged Referrals in advance, through a Clinical Genetics department if possible. Patients presenting with a phenotype appropriate for the requested sub-panel Amniotic fluid or CV samples Referrals will be accepted from clinical geneticists and consultants in ophthalmology. should be sent to Cytogenetics for Prenatal testing dissecting and culturing, with instructions to forward the sample Prenatal diagnosis may be offered as appropriate where pathogenic variants have been to the Regional Molecular Genetics identified in accordance with expected inheritance pattern and where appropriate parental laboratory for analysis testing and counselling has been conducted. -

17-2021 CAMI Pilot Vision Brochure
Visual Scanning with regular eye examinations and post surgically with phoria results. A pilot who has such a condition could progress considered for medical certification through special issuance with Some images used from The Federal Aviation Administration. monofocal lenses when they meet vision standards without to seeing double (tropia) should they be exposed to hypoxia or a satisfactory adaption period, complete evaluation by an eye Helicopter Flying Handbook. Oklahoma City, Ok: US Department The probability of spotting a potential collision threat complications. Multifocal lenses require a brief waiting certain medications. specialist, satisfactory visual acuity corrected to 20/20 or better by of Transportation; 2012; 13-1. Publication FAA-H-8083. Available increases with the time spent looking outside, but certain period. The visual effects of cataracts can be successfully lenses of no greater power than ±3.5 diopters spherical equivalent, at: https://www.faa.gov/regulations_policies/handbooks_manuals/ techniques may be used to increase the effectiveness of treated with a 90% improvement in visual function for most One prism diopter of hyperphoria, six prism diopters of and by passing an FAA medical flight test (MFT). aviation/helicopter_flying_handbook/. Accessed September 28, 2017. the scan time. Effective scanning is accomplished with a patients. Regardless of vision correction to 20/20, cataracts esophoria, and six prism diopters of exophoria represent series of short, regularly-spaced eye movements that bring pose a significant risk to flight safety. FAA phoria (deviation of the eye) standards that may not be A Word about Contact Lenses successive areas of the sky into the central visual field. Each exceeded. -

Transgenic Zebrafish Models Reveal Distinct Molecular Mechanisms for Cataract-Linked Αa-Crystallin Mutants
bioRxiv preprint doi: https://doi.org/10.1101/364125; this version posted July 8, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Transgenic Zebrafish Models Reveal Distinct Molecular Mechanisms for Cataract-linked αA-Crystallin Mutants Shu-Yu Wu, Ping Zou, Sanjay Mishra, Hassane S Mchaourab* Department of Molecular Physiology and Biophysics, Vanderbilt University, Nashville, TN 37232, USA Running title: Distinct mechanisms of α-crystallin mutations * Corresponding author: Hassane Mchaourab 741 Light Hall 2215 Garland Avenue Molecular Physiology & Biophysics Nashville, TN 37232 Office: 615.322.3307 Fax: 615.322.7236 Email: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/364125; this version posted July 8, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract Mutations in the small heat shock proteins a-crystallins have been linked to autosomal dominant cataracts in humans. Extensive studies in vitro have revealed a spectrum of alterations to the structure and function of these proteins including shifts in the size of the oligomer, modulation of subunit exchange and modification of their affinity to client proteins. Although mouse models of these mutants were instrumental in identifying changes in cellular proliferation and lens development, a direct comparative analysis of their effects on lens proteostasis has not been performed. -

Causes of Color Blindness: Function and Failure of the Genes That Detect Color
A REVIEW ON COLOR BLINDNESS 1 Causes of Color Blindness: Function and Failure of the Genes that Detect Color Dylan Taylor A Senior Thesis submitted in partial fulfillment of the requirements for graduation in the Honors Program Liberty University Fall 2020 A REVIEW ON COLOR BLINDNESS 2 Acceptance of Senior Honors Thesis This Senior Honors Thesis is accepted in partial fulfillment of the requirements for graduation from the Honors Program of Liberty University. ______________________________ Gary D. Isaacs Jr., Ph.D. Thesis Chair ______________________________ Michael S. Price, Ph.D. Committee Member _____________________________ James H. Nutter, D.A. Honors Director ______________________________ Date A REVIEW ON COLOR BLINDNESS 3 Abstract Color blindness affects nearly 10% of the entire population, with multiple types of color blindness from various genetic mutations. In the following sections, the nature of light and how the human eye perceives light will be discussed. Afterward, the major forms of color blindness and their genetic causes will be considered. Once these genetic causes have been established, the current method for diagnosing color blindness will be investigated, followed by a discussion of the current treatments available to those with color blindness. Finally, a brief discussion will address possible future work for color blindness with the hope of finding better treatments and a future prevention. A REVIEW ON COLOR BLINDNESS 4 Causes of Color Blindness: Function and Failure of the Genes that Detect Color Introduction Without the human eye’s ability to detect color, the world would appear as dull as a black and white movie. However, despite our ability to detect color, not all humans perceive the same colors as one another. -

The Visual Ecology of Avian Photoreceptors Nathan S
PII: S1350-9462(01)00009-X The Visual Ecology of Avian Photoreceptors Nathan S. Hart* Vision, Touch and Hearing Research Centre, Department of Physiology and Pharmacology, The University of Queensland, Brisbane 4072, Australia CONTENTS Abstract . 676 1. Introduction . 676 1.1. The avian visual system . 676 1.2. Microspectrophotometry and the study ofavian vision . 676 2. Avian retinal photoreceptors . 681 2.1. Visual pigments . 681 2.2. Cone oil droplets . 684 3. UVS/VS cones . 685 3.1. Microspectrophotometric data . 685 3.2. Spectral tuning and phylogeny . 685 3.3. Evolution ofSWS1 opsin-based visual pigments in birds . 686 3.4. The short wavelength limit ofphotoreception . 686 4. SWS single cones. 687 4.1. Microspectrophotometric data . 687 4.2. Dependence upon UVS/VS single cone spectral sensitivity . 688 4.3. Spectral tuning . 688 5. Rods and MWS single cones . 688 5.1. Comparison ofavian RH1 and RH2 opsins . 688 5.2. The duplex retina . 689 5.3. Factors influencing rod visual pigment kmax ........................ 689 6. LWS single and double cones . 691 6.1. Microspectrophotometric data . 691 6.2. Spectral tuning . 691 6.3. Effect ofoil droplet transmittance on spectral sensitivity . 692 6.4. Double cones and the interspecific variation in LWS visual pigment kmax ........ 692 6.5. Spectral filtering by double cone oil droplets . 694 7. Variations in the relative proportions ofdifferent cone types . 695 7.1. Interspecific variations . 695 7.2. Intraretinal variations . 696 7.3. Bilateral asymmetry . 697 8. Conclusions . 697 9. Future directions . 698 Acknowledgements . 698 References . 698 *Tel.: +61-7-3365-1867; fax: +61-7-3365-4522; e-mail: [email protected]. -

Rescue of Retinal Degeneration by Intravitreally Injected Adult Bone Marrow–Derived Lineage-Negative Hematopoietic Stem Cells
Rescue of retinal degeneration by intravitreally injected adult bone marrow–derived lineage-negative hematopoietic stem cells Atsushi Otani, … , John Heckenlively, Martin Friedlander J Clin Invest. 2004;114(6):765-774. https://doi.org/10.1172/JCI21686. Article Stem cells Inherited retinal degenerations afflict 1 in 3,500 individuals and are a heterogeneous group of diseases that result in profound vision loss, usually the result of retinal neuronal apoptosis. Atrophic changes in the retinal vasculature are also observed in many of these degenerations. While it is thought that this atrophy is secondary to diminished metabolic demand in the face of retinal degeneration, the precise relationship between the retinal neuronal and vascular degeneration is not clear. In this study we demonstrate that whenever a fraction of mouse or human adult bone marrow– derived stem cells (lineage-negative hematopoietic stem cells [Lin– HSCs]) containing endothelial precursors stabilizes and rescues retinal blood vessels that would ordinarily completely degenerate, a dramatic neurotrophic rescue effect is also observed. Retinal nuclear layers are preserved in 2 mouse models of retinal degeneration, rd1 and rd10, and detectable, albeit severely abnormal, electroretinogram recordings are observed in rescued mice at times when they are never observed in control-treated or untreated eyes. The normal mouse retina consists predominantly of rods, but the rescued cells after treatment with Lin– HSCs are nearly all cones. Microarray analysis of rescued retinas demonstrates