User's Guide for Quantum ESPRESSO
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Using GNU Fortran 95
Contributed by Steven Bosscher ([email protected]). Using GNU Fortran 95 Steven Bosscher For the 4.0.4 Version* Published by the Free Software Foundation 59 Temple Place - Suite 330 Boston, MA 02111-1307, USA Copyright c 1999-2005 Free Software Foundation, Inc. Permission is granted to copy, distribute and/or modify this document under the terms of the GNU Free Documentation License, Version 1.1 or any later version published by the Free Software Foundation; with the Invariant Sections being “GNU General Public License” and “Funding Free Software”, the Front-Cover texts being (a) (see below), and with the Back-Cover Texts being (b) (see below). A copy of the license is included in the section entitled “GNU Free Documentation License”. (a) The FSF’s Front-Cover Text is: A GNU Manual (b) The FSF’s Back-Cover Text is: You have freedom to copy and modify this GNU Manual, like GNU software. Copies published by the Free Software Foundation raise funds for GNU development. i Short Contents Introduction ...................................... 1 GNU GENERAL PUBLIC LICENSE .................... 3 GNU Free Documentation License ....................... 9 Funding Free Software .............................. 17 1 Getting Started................................ 19 2 GFORTRAN and GCC .......................... 21 3 GFORTRAN and G77 ........................... 23 4 GNU Fortran 95 Command Options.................. 25 5 Project Status ................................ 33 6 Extensions ................................... 37 7 Intrinsic Procedures ............................ -

User's Guide for Quantum ESPRESSO
User's Guide for Quantum ESPRESSO (v.6.1) Contents 1 Introduction 1 1.1 People . 3 1.2 Contacts . 4 1.3 Guidelines for posting to the mailing list . 4 1.4 Terms of use . 5 2 Installation 5 2.1 Download . 6 2.2 Prerequisites . 7 2.3 configure ....................................... 7 2.3.1 Manual configuration . 9 2.4 Libraries . 10 2.5 Compilation . 11 2.6 Running tests and examples . 12 2.7 Installation tricks and problems . 14 2.7.1 All architectures . 14 2.7.2 Intel Xeon Phi . 15 2.7.3 Cray machines . 15 2.7.4 IBM AIX . 16 2.7.5 IBM BlueGene . 16 2.7.6 Linux PC . 16 2.7.7 Linux PC clusters with MPI . 18 2.7.8 Mac OS . 19 3 Parallelism 21 3.1 Understanding Parallelism . 21 3.2 Running on parallel machines . 21 3.3 Parallelization levels . 22 3.3.1 Understanding parallel I/O . 24 3.4 Tricks and problems . 24 1 1 Introduction This guide gives a general overview of the contents and of the installation of Quantum ESPRESSO (opEn-Source Package for Research in Electronic Structure, Simulation, and Op- timization), version 6.1. The Quantum ESPRESSO distribution contains the core packages PWscf (Plane-Wave Self-Consistent Field) and CP (Car-Parrinello) for the calculation of electronic-structure prop- erties within Density-Functional Theory (DFT), using a Plane-Wave (PW) basis set and pseu- dopotentials. It also includes other packages for more specialized calculations: • PWneb: energy barriers and reaction pathways through the Nudged Elastic Band (NEB) method. -

Coarrays in GNU Fortran
Coarrays in GNU Fortran Alessandro Fanfarillo [email protected] June 24th, 2014 Alessandro Fanfarillo Coarrays in GFortran June 24th, 2014 1 / 50 Introduction Coarray Fortran (also known as CAF) is a syntactic extension of Fortran 95/2003 which has been included in the Fortran 2008 standard. The main goal is to allow Fortran users to realize parallel programs without the burden of explicitly invoke communication functions or directives (MPI, OpenMP). The secondary goal is to express parallelism in a \platform-agnostic" way (no explicit shared or distributed paradigm). Coarrays are based on the Partitioned Global Address Space model (PGAS). Compilers which support Coarrays: Cray Compiler (Gold standard - Commercial) Intel Compiler (Commercial) Rice Compiler (Free - Rice University) OpenUH (Free - University of Houston) G95 (Coarray support not totally free - Not up to date) Alessandro Fanfarillo Coarrays in GFortran June 24th, 2014 2 / 50 PGAS Languages The PGAS model assumes a global memory address space that is logically partitioned and a portion of it is local to each process or thread. It means that a process can directly access a memory portion owned by another process. The model attempts to combine the advantages of a SPMD programming style for distributed memory systems (as employed by MPI) with the data referencing semantics of shared memory systems. Coarray Fortran. UPC (upc.gwu.edu). Titanium (titanium.cs.berkeley.edu). Chapel (Cray). X10 (IBM). Alessandro Fanfarillo Coarrays in GFortran June 24th, 2014 3 / 50 Coarray concepts A program is treated as if it were replicated at the start of execution, each replication is called an image. -

Pathscale Fortran and C/C++ Compilers
PathScale™ Compiler Suite User Guide Version 3.2 Page i PathScale Compiler Suite User Guide Version 3.2 Page ii PathScale Compiler Suite User Guide Version 3.2 Information furnished in this manual is believed to be accurate and reliable. However, PathScale LLC assumes no responsibility for its use, nor for any infringements of patents or other rights of third parties which may result from its use. PathScale LLC reserves the right to change product specifications at any time without notice. Applications described in this document for any of these products are for illustrative purposes only. PathScale LLC makes no representation nor warranty that such applications are suitable for the specified use without further testing or modification. PathScale LLC assumes no responsibility for any errors that may appear in this document. No part of this document may be copied nor reproduced by any means, nor translated nor transmitted to any magnetic medium without the express written consent of PathScale LLC. In accordance with the terms of their valid PathScale agreements, customers are permitted to make electronic and paper copies of this document for their own exclusive use. Linux is a registered trademark of Linus Torvalds. PathScale, the PathScale logo, and EKOPath are registered trademarks of PathScale, LLC. Red Hat and all Red Hat-based trademarks are trademarks or registered trademarks of Red Hat, Inc. SuSE is a registered trademark of SuSE Linux AG. All other brand and product names are trademarks or registered trademarks of their respective owners. © 2007, 2008 PathScale, LLC. All rights reserved. © 2006, 2007 QLogic Corporation. All rights reserved worldwide. -

Using and Porting GNU Fortran
Contributed by James Craig Burley ([email protected]). Inspired by a first pass at translating ‘g77-0.5.16/f/DOC’ that was contributed to Craig by David Ronis ([email protected]). Using and Porting GNU Fortran James Craig Burley Last updated 2004-03-21 for version GCC-3.4.6 For the GCC-3.4.6 Version* Published by the Free Software Foundation 59 Temple Place - Suite 330 Boston, MA 02111-1307, USA Copyright c 1995,1996,1997,1998,1999,2000,2001,2002,2003,2004 Free Software Founda- tion, Inc. Permission is granted to copy, distribute and/or modify this document under the terms of the GNU Free Documentation License, Version 1.2 or any later version published by the Free Software Foundation; with the Invariant Sections being “GNU General Public License” and “Funding Free Software”, the Front-Cover texts being (a) (see below), and with the Back-Cover Texts being (b) (see below). A copy of the license is included in the section entitled “GNU Free Documentation License”. (a) The FSF’s Front-Cover Text is: A GNU Manual (b) The FSF’s Back-Cover Text is: You have freedom to copy and modify this GNU Manual, like GNU software. Copies published by the Free Software Foundation raise funds for GNU development. i Short Contents Introduction ...................................... 1 GNU GENERAL PUBLIC LICENSE .................... 3 GNU Free Documentation License ....................... 9 Contributors to GNU Fortran ......................... 17 Funding Free Software .............................. 19 1 Funding GNU Fortran ........................... 21 2 Getting Started................................ 23 3 What is GNU Fortran? .......................... 25 4 Compile Fortran, C, or Other Programs .............. -

Programming with GNU Software
1 Programming with GNU Software Edition 2, 4 September 2002 $Id: gnuprog2.texi,v 1.22 2002/08/15 19:41:25 rmeeking Exp $ 2 Programming with GNU Software Copyright c 2000, 2001 Gary V. Vaughan, Akim Demaille, Paul Scott, Bruce Korb, Richard Meeking Permission is granted to copy, distribute and/or modify this document under the terms of the GNU Free Documentation License, Version 1.1 or any later version published by the Free Software Foundation; with the no Invariant Sections, with no Front-Cover Texts, and with no Back-Cover Texts. A copy of the license is included in the section entitled "GNU Free Documentation License". i Table of Contents Foreword . 1 1 Introduction . 3 1.1 what this book is . 3 1.2 What the gde is not. 3 1.3 Audience . 4 1.4 this book's organization. 4 1.5 What You Are Presumed to Know . 4 1.6 Conventions Used in This Book. 6 2 The GNU C Library. 7 2.1 Glibc Architecture Overview . 7 2.2 Standards Conformance. 9 2.3 Memory Management. 9 2.3.1 alloca . 9 2.3.2 obstack . 11 2.3.3 argz . 14 2.4 Input and Output . 14 2.4.1 Signals . 14 2.4.2 Time Formats . 17 2.4.3 Formatted Printing . 22 2.5 Error Handling. 22 2.6 Pattern Matching . 24 2.6.1 Wildcard Matching . 24 2.6.2 Filename Matching . 27 2.6.3 Regular Expression Matching . 27 3 libstdc++ and the Standard Template Library . 29 3.1 How the STL is Structured. -
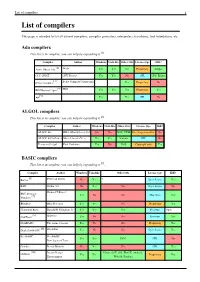
List of Compilers 1 List of Compilers
List of compilers 1 List of compilers This page is intended to list all current compilers, compiler generators, interpreters, translators, tool foundations, etc. Ada compilers This list is incomplete; you can help by expanding it [1]. Compiler Author Windows Unix-like Other OSs License type IDE? [2] Aonix Object Ada Atego Yes Yes Yes Proprietary Eclipse GCC GNAT GNU Project Yes Yes No GPL GPS, Eclipse [3] Irvine Compiler Irvine Compiler Corporation Yes Proprietary No [4] IBM Rational Apex IBM Yes Yes Yes Proprietary Yes [5] A# Yes Yes GPL No ALGOL compilers This list is incomplete; you can help by expanding it [1]. Compiler Author Windows Unix-like Other OSs License type IDE? ALGOL 60 RHA (Minisystems) Ltd No No DOS, CP/M Free for personal use No ALGOL 68G (Genie) Marcel van der Veer Yes Yes Various GPL No Persistent S-algol Paul Cockshott Yes No DOS Copyright only Yes BASIC compilers This list is incomplete; you can help by expanding it [1]. Compiler Author Windows Unix-like Other OSs License type IDE? [6] BaCon Peter van Eerten No Yes ? Open Source Yes BAIL Studio 403 No Yes No Open Source No BBC Basic for Richard T Russel [7] Yes No No Shareware Yes Windows BlitzMax Blitz Research Yes Yes No Proprietary Yes Chipmunk Basic Ronald H. Nicholson, Jr. Yes Yes Yes Freeware Open [8] CoolBasic Spywave Yes No No Freeware Yes DarkBASIC The Game Creators Yes No No Proprietary Yes [9] DoyleSoft BASIC DoyleSoft Yes No No Open Source Yes FreeBASIC FreeBASIC Yes Yes DOS GPL No Development Team Gambas Benoît Minisini No Yes No GPL Yes [10] Dream Design Linux, OSX, iOS, WinCE, Android, GLBasic Yes Yes Proprietary Yes Entertainment WebOS, Pandora List of compilers 2 [11] Just BASIC Shoptalk Systems Yes No No Freeware Yes [12] KBasic KBasic Software Yes Yes No Open source Yes Liberty BASIC Shoptalk Systems Yes No No Proprietary Yes [13] [14] Creative Maximite MMBasic Geoff Graham Yes No Maximite,PIC32 Commons EDIT [15] NBasic SylvaWare Yes No No Freeware No PowerBASIC PowerBASIC, Inc. -

A Coarray Fortran Implementation to Support Data-Intensive Application Development
A Coarray Fortran implementation to support data-intensive application development The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Eachempati, Deepak, Alan Richardson, Siddhartha Jana, Terrence Liao, Henri Calandra, and Barbara Chapman. “A Coarray Fortran Implementation to Support Data-Intensive Application Development.” Cluster Comput 17, no. 2 (October 12, 2013): 569– 583. As Published http://dx.doi.org/10.1007/s10586-013-0302-7 Publisher Springer US Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/104091 Terms of Use Article is made available in accordance with the publisher's policy and may be subject to US copyright law. Please refer to the publisher's site for terms of use. CLUS302_source [09/04 13:33] 1/12 A Coarray Fortran Implementation to Support Data-Intensive Application Development Deepak Eachempati∗, Alan Richardsony, Siddhartha Jana∗, Terrence Liaoz, Henri Calandrax and Barbara Chapman∗ ∗Department of Computer Science, University of Houston, Houston, TX Email: fdreachem,sidjana,[email protected] yDepartment of Earth, Atmospheric, and Planetary Sciences (EAPS), MIT, Cambridge, MA Email: alan [email protected] zTOTAL E&P R&T USA, LLC, Houston, TX Email: [email protected] xTOTAL E&P, Pau, France Email: [email protected] Abstract—In this paper, we describe our experiences in im- at which data is input and output from and to disk. In order plementing and applying Coarray Fortran (CAF) for the devel- to accommodate it, we believe that there is a need for parallel opment of data-intensive applications in the domain of Oil and I/O features in Fortran which complement the existing parallel- Gas exploration. -

Opencoarrays: Open-Source Transport Layers Supporting Coarray Fortran Compilers
OpenCoarrays: Open-source Transport Layers Supporting Coarray Fortran Compilers ∗ y z Alessandro Fanfarillo Tobias Burnus Valeria Cardellini University of Rome Munich, Germany University of Rome Tor Vergata Tor Vergata Rome, Italy Rome, Italy x { q Salvatore Filippone Dan Nagle Damian Rouson University of Rome National Center for Sourcery Inc. Tor Vergata Atmospheric Research Oakland, California Rome, Italy Boulder, Colorado ABSTRACT 1. INTRODUCTION Coarray Fortran is a set of features of the Fortran 2008 stan- Coarray Fortran (also known as CAF) originated as a syn- dard that make Fortran a PGAS parallel programming lan- tactic extension of Fortran 95 proposed in the early 1990s guage. Two commercial compilers currently support coar- by Robert Numrich and John Reid [10] and eventually be- rays: Cray and Intel. Here we present two coarray trans- came part of the Fortran 2008 standard published in 2010 port layers provided by the new OpenCoarrays project: one (ISO/IEC 1539-1:2010) [11]. The main goal of coarrays is library based on MPI and the other on GASNet. We link to allow Fortran users to create parallel programs without the GNU Fortran (GFortran) compiler to either of the two the burden of explicitly invoking communication functions OpenCoarrays implementations and present performance com- or directives such as those in the Message Passing Interface parisons between executables produced by GFortran and the (MPI) [12] and OpenMP [2]. Cray and Intel compilers. The comparison includes syn- thetic benchmarks, application prototypes, and an applica- Coarrays are based on the Partitioned Global Address Space tion kernel. In our tests, Intel outperforms GFortran only on (PGAS) parallel programming model, which attempts to intra-node small transfers (in particular, scalars). -

A Coarray Fortran Implementation to Support Data-Intensive Application Development
A Coarray Fortran Implementation to Support Data-Intensive Application Development Deepak Eachempati∗, Alan Richardsony, Terrence Liaoz, Henri Calandrax and Barbara Chapman∗ ∗Department of Computer Science, University of Houston, Houston, TX Email: fdreachem,[email protected] yDepartment of Earth, Atmospheric, and Planetary Sciences (EAPS), MIT, Cambridge, MA Email: alan [email protected] zTOTAL E&P R&T USA, LLC, Houston, TX Email: [email protected] xTOTAL E&P, Pau, France Email: [email protected] Abstract—We describe our experiences in implementing and conclusions in section VII. applying the Coarray Fortran language extensions adopted in Fortran 2008. The successful porting of reverse time migration II. COARRAY FORTRAN (RTM), a data-intensive algorithm and one of the largest uses of The CAF extension in Fortran 2008 adds new features to computational resources in seismic exploration, is described, and results are presented demonstrating that the CAF implementation the language for parallel programming. It follows the Single- provides comparable performance to an equivalent MPI version. Program, Multiple-Data (SPMD) programming model, where We close with a discussion on parallel I/O and how it may be multiple copies of the program, termed images in CAF, execute incorporated into the CAF programming model. asynchronously and have local data objects that are globally accessible. CAF may be implemented on top of shared mem- I. INTRODUCTION ory systems and distributed memory systems. The programmer New programming models are needed to meet the chal- may access memory of any remote image without the explicit lenges posed by ever-increasing data processing requirements. involvement of that image, as if the data is “distributed” across They must provide mechanisms for accessing and processing all the images and there is a single, global address space large-scale data sets. -

Fortran Resources1
Fortran Resources1 Ian D Chivers Jane Sleightholme September 16, 2008 1The original basis for this document was Mike Metcalf’s Fortran Information File. The next input came from people on comp-fortran-90. Details of how to subscribe or browse this list can be found in this document. If you have any corrections, additions, suggestions etc to make please contact us and we will endeavor to include your comments in later versions. Thanks to all the people who have contributed. 2 Contents 1 Fortran 90, 95 and 2003 Books 9 1.1 Fortran 2003 - English . 9 1.2 Fortran 95 - English . 9 1.3 Fortran 90 - English . 10 1.4 English books on related topics . 11 1.5 Chinese . 12 1.6 Dutch . 12 1.7 Finnish . 12 1.8 French . 12 1.9 German . 13 1.10 Italian . 14 1.11 Japanese . 14 1.12 Russian . 14 1.13 Swedish . 14 2 Fortran 90, 95 and 2003 Compilers 15 2.1 Introduction . 15 2.2 Absoft . 15 2.3 Apogee . 16 2.4 Compaq . 16 2.5 Cray . 16 2.6 Fortran Company . 16 2.7 Fujitsu . 17 2.8 Gnu Fortran 95 . 17 2.9 G95 . 18 2.10 Hewlett Packard . 18 2.11 IBM . 19 2.12 Intel . 19 2.13 Lahey/Fujitsu . 20 2.14 NAG . 21 2.15 NA Software . 21 2.16 NEC . 22 2.17 PathScale . 22 2.18 PGI . 22 2.19 Salford Software . 23 4 CONTENTS 2.20 SGI . 23 2.21 Sun . 23 3 Fortran aware editors or development environments 25 3.1 Windows . -

Using GNU Fortran 95
Using GNU Fortran 95 The gfortran team For the 4.1.2 Version* Published by the Free Software Foundation 51 Franklin Street, Fifth Floor Boston, MA 02110-1301, USA Copyright c 1999-2005 Free Software Foundation, Inc. Permission is granted to copy, distribute and/or modify this document under the terms of the GNU Free Documentation License, Version 1.1 or any later version published by the Free Software Foundation; with the Invariant Sections being “GNU General Public License” and “Funding Free Software”, the Front-Cover texts being (a) (see below), and with the Back-Cover Texts being (b) (see below). A copy of the license is included in the section entitled “GNU Free Documentation License”. (a) The FSF’s Front-Cover Text is: A GNU Manual (b) The FSF’s Back-Cover Text is: You have freedom to copy and modify this GNU Manual, like GNU software. Copies published by the Free Software Foundation raise funds for GNU development. i Short Contents Introduction ...................................... 1 1 Getting Started ................................ 3 2 GFORTRAN and GCC ........................... 5 3 GFORTRAN and G77 ............................ 7 4 GNU Fortran 95 Command Options .................. 9 5 Project Status ................................ 19 6 Runtime: Influencing runtime behavior with environment variables .................................... 23 7 Extensions ................................... 27 8 Intrinsic Procedures ............................. 33 9 Contributing.................................. 89 10 Standards ................................... 91 GNU GENERAL PUBLIC LICENSE .................... 93 GNU Free Documentation License ...................... 99 Funding Free Software ............................. 107 Index ......................................... 109 ii The GNU Fortran 95 Compiler iii Table of Contents Introduction .................................. 1 1 Getting Started............................ 3 2 GFORTRAN and GCC .................... 5 3 GFORTRAN and G77 ..................... 7 4 GNU Fortran 95 Command Options .......