SCIENCE CHINA Cytoplasmic Dynein
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Unsheathing WASP's Sting
news and views required for Swallow-mediated localiza- Cytoplasmic dynein is implicated in Minneapolis 55455, Minnesota, USA tion within the oocyte. many biological processes, including ves- e-mail:[email protected] The interaction between Swallow and icle and organelle transport, mitotic- Roger Karess is at the CNRS Centre de Génétique Dlc is a significant finding and provides spindle function and orientation, and Moléculaire, Ave de la Terrasse, 91198 Gif-sur- the basis for a model in which the dynein- now RNA transport and localization. It is Yvette, France motor complex is responsible for the important to emphasize that a single iso- e-mail: [email protected] anterior localization of bicoid RNA within form of the dynein-motor subunit is 1. St Johnston, D. Cell 81, 167–170 (1995). the oocyte. Interestingly, the transient known to be targeted to several cellular 2. Bashirullah, A., Cooperstock, R. & Lipshitz, H. Annu. Rev. localization of Swallow to the oocyte functions and molecular cargoes within Biochem. 67, 335–394 (1998). anterior occurs at a time when most of the individual cells. Thus it is those molecules 3. Oleynikov, Y. & Singer, R. Trends Cell Biol. 8, 381–383 dynein-motor subunits are concentrated with adaptor functions, such as those pro- (1998). 13 4. Wilhelm, J. & Vale, R. J. Cell Biol. 123, 269–274 (1993). at the posterior of the oocyte . This raises posed here for Swallow, that must 5. Schnorrer, F., Bohmann, K. & Nusslein-Volhard, C. Nature Cell the possibility that at least two distinct account for the functional specificity of Biol. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Tracking Melanosomes Inside a Cell to Study Molecular Motors and Their Interaction
Tracking melanosomes inside a cell to study molecular motors and their interaction Comert Kural*, Anna S. Serpinskaya†, Ying-Hao Chou†, Robert D. Goldman†, Vladimir I. Gelfand†‡, and Paul R. Selvin*§¶ *Center for Biophysics and Computational Biology and §Department of Physics, University of Illinois at Urbana–Champaign, Urbana, IL 61801; and †Department of Cell and Molecular Biology, Northwestern University School of Medicine, Chicago, IL 60611 Communicated by Gordon A. Baym, University of Illinois at Urbana–Champaign, Urbana, IL, January 9, 2007 (received for review June 4, 2006) Cells known as melanophores contain melanosomes, which are membrane organelles filled with melanin, a dark, nonfluorescent pigment. Melanophores aggregate or disperse their melanosomes when the host needs to change its color in response to the environment (e.g., camouflage or social interactions). Melanosome transport in cultured Xenopus melanophores is mediated by my- osin V, heterotrimeric kinesin-2, and cytoplasmic dynein. Here, we describe a technique for tracking individual motors of each type, both individually and in their interaction, with high spatial (Ϸ2 nm) and temporal (Ϸ1 msec) localization accuracy. This method enabled us to observe (i) stepwise movement of kinesin-2 with an average step size of 8 nm; (ii) smoother melanosome transport (with fewer pauses), in the absence of intermediate filaments (IFs); and (iii) motors of actin filaments and microtubules working on the same cargo nearly simultaneously, indicating that a diffusive step is not needed between the two systems of transport. In concert with our previous report, our results also show that dynein-driven retro- grade movement occurs in 8-nm steps. Furthermore, previous studies have shown that melanosomes carried by myosin V move 35 nm in a stepwise fashion in which the step rise-times can be as long as 80 msec. -

Cytoplasmic Dynein Pushes the Cytoskeletal Meshwork Forward During Axonal Elongation
ß 2014. Published by The Company of Biologists Ltd | Journal of Cell Science (2014) 127, 3593–3602 doi:10.1242/jcs.152611 RESEARCH ARTICLE Cytoplasmic dynein pushes the cytoskeletal meshwork forward during axonal elongation Douglas H. Roossien1, Phillip Lamoureux2 and Kyle E. Miller2,* ABSTRACT were not a species-specific phenomenon, but rather a broadly conserved mechanism for elongation. From this, a new model for During development, neurons send out axonal processes that can axonal elongation has emerged, termed ‘stretch and intercalation’ reach lengths hundreds of times longer than the diameter of their (SAI) (Suter and Miller, 2011), in which forces cause the cell bodies. Recent studies indicate that en masse microtubule microtubule-rich central domain (C-domain) of the growth cone translocation is a significant mechanism underlying axonal to advance in bulk. This is paired with stretching of the axon, elongation, but how cellular forces drive this process is unknown. which is followed by intercalated mass addition along the axon Cytoplasmic dynein generates forces on microtubules in axons to to prevent thinning (Lamoureux et al., 2010). In terms of the power their movement through ‘stop-and-go’ transport, but whether cytoskeleton, stretching presumably occurs because filaments are these forces influence the bulk translocation of long microtubules sliding apart either through pulling or pushing forces generated embedded in the cytoskeletal meshwork has not been tested. by molecular motors (Suter and Miller, 2011; Lu et al., 2013; Here, we use both function-blocking antibodies targeted to Roossien et al., 2013). It is worth noting that because adhesions the dynein intermediate chain and the pharmacological dynein along the axon dissipate forces generated in the growth inhibitor ciliobrevin D to ask whether dynein forces contribute to en cone (O’Toole et al., 2008), these en masse movements of bloc cytoskeleton translocation. -

Proteomic and Bioinformatic Investigation of Altered Pathways in Neuroglobin-Deficient Breast Cancer Cells
molecules Article Proteomic and Bioinformatic Investigation of Altered Pathways in Neuroglobin-Deficient Breast Cancer Cells Michele Costanzo 1,2 , Marco Fiocchetti 3 , Paolo Ascenzi 3, Maria Marino 3 , Marianna Caterino 1,2,* and Margherita Ruoppolo 1,2,* 1 Department of Molecular Medicine and Medical Biotechnology, School of Medicine, University of Naples Federico II, 80131 Naples, Italy; [email protected] 2 CEINGE—Biotecnologie Avanzate S.C.Ar.L., 80145 Naples, Italy 3 Department of Science, University Roma Tre, 00146 Rome, Italy; marco.fi[email protected] (M.F.); [email protected] (P.A.); [email protected] (M.M.) * Correspondence: [email protected] (M.C.); [email protected] (M.R.) Abstract: Neuroglobin (NGB) is a myoglobin-like monomeric globin that is involved in several processes, displaying a pivotal redox-dependent protective role in neuronal and extra-neuronal cells. NGB remarkably exerts its function upon upregulation by NGB inducers, such as 17β-estradiol (E2) and H2O2. However, the molecular bases of NGB’s functions remain undefined, mainly in non- neuronal cancer cells. Human MCF-7 breast cancer cells with a knocked-out (KO) NGB gene obtained using CRISPR/Cas9 technology were analyzed using shotgun label-free quantitative proteomics in comparison with control cells. The differential proteomics experiments were also performed after treatment with E2, H2O2, and E2 + H2O2. All the runs acquired using liquid chromatography–tandem mass spectrometry were elaborated within the same MaxQuant analysis, leading to the quantification Citation: Costanzo, M.; Fiocchetti, of 1872 proteins in the global proteomic dataset. Then, a differentially regulated protein dataset was M.; Ascenzi, P.; Marino, M.; Caterino, M.; Ruoppolo, M. -

Tension on the Linker Gates the ATP-Dependent Release of Dynein from Microtubules
ARTICLE Received 4 Apr 2014 | Accepted 3 Jul 2014 | Published 11 Aug 2014 DOI: 10.1038/ncomms5587 Tension on the linker gates the ATP-dependent release of dynein from microtubules Frank B. Cleary1, Mark A. Dewitt1, Thomas Bilyard2, Zaw Min Htet2, Vladislav Belyy1, Danna D. Chan2, Amy Y. Chang2 & Ahmet Yildiz2 Cytoplasmic dynein is a dimeric motor that transports intracellular cargoes towards the minus end of microtubules (MTs). In contrast to other processive motors, stepping of the dynein motor domains (heads) is not precisely coordinated. Therefore, the mechanism of dynein processivity remains unclear. Here, by engineering the mechanical and catalytic properties of the motor, we show that dynein processivity minimally requires a single active head and a second inert MT-binding domain. Processivity arises from a high ratio of MT-bound to unbound time, and not from interhead communication. In addition, nucleotide-dependent microtubule release is gated by tension on the linker domain. Intramolecular tension sensing is observed in dynein’s stepping motion at high interhead separations. On the basis of these results, we propose a quantitative model for the stepping characteristics of dynein and its response to chemical and mechanical perturbation. 1 Biophysics Graduate Group, University of California, Berkeley, California 94720, USA. 2 Department of Physics, University of California, Berkeley, California 94720, USA. Correspondence and requests for materials should be addressed to A.Y. (email: [email protected]). NATURE COMMUNICATIONS | 5:4587 | DOI: 10.1038/ncomms5587 | www.nature.com/naturecommunications 1 & 2014 Macmillan Publishers Limited. All rights reserved. ARTICLE NATURE COMMUNICATIONS | DOI: 10.1038/ncomms5587 ytoplasmic dynein is responsible for nearly all microtubule dynein’s MT-binding domain (MTBD) is located at the end of a (MT) minus-end-directed transport in eukaryotes1.In coiled-coil stalk10. -

Supplementary Table S1. Correlation Between the Mutant P53-Interacting Partners and PTTG3P, PTTG1 and PTTG2, Based on Data from Starbase V3.0 Database
Supplementary Table S1. Correlation between the mutant p53-interacting partners and PTTG3P, PTTG1 and PTTG2, based on data from StarBase v3.0 database. PTTG3P PTTG1 PTTG2 Gene ID Coefficient-R p-value Coefficient-R p-value Coefficient-R p-value NF-YA ENSG00000001167 −0.077 8.59e-2 −0.210 2.09e-6 −0.122 6.23e-3 NF-YB ENSG00000120837 0.176 7.12e-5 0.227 2.82e-7 0.094 3.59e-2 NF-YC ENSG00000066136 0.124 5.45e-3 0.124 5.40e-3 0.051 2.51e-1 Sp1 ENSG00000185591 −0.014 7.50e-1 −0.201 5.82e-6 −0.072 1.07e-1 Ets-1 ENSG00000134954 −0.096 3.14e-2 −0.257 4.83e-9 0.034 4.46e-1 VDR ENSG00000111424 −0.091 4.10e-2 −0.216 1.03e-6 0.014 7.48e-1 SREBP-2 ENSG00000198911 −0.064 1.53e-1 −0.147 9.27e-4 −0.073 1.01e-1 TopBP1 ENSG00000163781 0.067 1.36e-1 0.051 2.57e-1 −0.020 6.57e-1 Pin1 ENSG00000127445 0.250 1.40e-8 0.571 9.56e-45 0.187 2.52e-5 MRE11 ENSG00000020922 0.063 1.56e-1 −0.007 8.81e-1 −0.024 5.93e-1 PML ENSG00000140464 0.072 1.05e-1 0.217 9.36e-7 0.166 1.85e-4 p63 ENSG00000073282 −0.120 7.04e-3 −0.283 1.08e-10 −0.198 7.71e-6 p73 ENSG00000078900 0.104 2.03e-2 0.258 4.67e-9 0.097 3.02e-2 Supplementary Table S2. -

Stable Tug-Of-War Between Kinesin-1 and Cytoplasmic Dynein Upon Different ATP and Roadblock Concentrations Gina A
© 2020. Published by The Company of Biologists Ltd | Journal of Cell Science (2020) 133, jcs249938. doi:10.1242/jcs.249938 RESEARCH ARTICLE Stable tug-of-war between kinesin-1 and cytoplasmic dynein upon different ATP and roadblock concentrations Gina A. Monzon1, Lara Scharrel2, Ashwin DSouza2, Verena Henrichs2,3, Ludger Santen1,* and Stefan Diez2,4,* ABSTRACT and dynein motors are known to be often simultaneously bound to The maintenance of intracellular processes, like organelle transport and the cargo (Gennerich and Schild, 2006; Welte, 2004; Soppina et al., cell division, depend on bidirectional movement along microtubules. 2009; Hendricks et al., 2010). Without any regulatory mechanism These processes typically require kinesin and dynein motor proteins, the cargo might be transported in the kinesin or dynein direction, which move with opposite directionality. Because both types of motors might randomly switch direction or might get stuck at a random are often simultaneously bound to the cargo, regulatory mechanisms are position. However, regulatory mechanisms ensuring targeted required to ensure controlled directional transport. Recently, it has transport remain poorly understood. been shown that parameters like mechanical motor activation, ATP In the past, different regulatory mechanisms have been proposed. concentration and roadblocks on the microtubule surface differentially One mechanism suggests coordinating the motor activity (Gross, influence the activity of kinesin and dynein motors in distinct manners. 2004). In this model, motors are assumed to be a priori in a passive However, how these parameters affect bidirectional transport state. By activating one motor team, targeted cargo transport occurs systems has not been studied. Here, we investigate the regulatory in the direction of the active team (Gross, 2004). -

Cytoskeleton Cytoskeleton
CYTOSKELETON CYTOSKELETON The cytoskeleton is composed of three principal types of protein filaments: actin filaments, intermediate filaments, and microtubules, which are held together and linked to subcellular organelles and the plasma membrane by a variety of accessory proteins Muscle Contraction • Skeletal muscles are bundles of muscle fibers • Most of the cytoplasm consists of myofibrils, which are cylindrical bundles of two types of filaments: thick filaments of myosin (about 15 run in diameter) and thin filaments of actin (about 7 nm in diameter). • Each myofibril is organized as a chain of contractile units called sarcomeres, which are responsible for the striated appearance of skeletal and cardiac muscle. Structure of muscle cells Sarcomere • The ends of each sarcomere are defined by the Z disc. • Within each sarcomere, dark bands (called A bands because they are anisotropic when viewed with polarized light) alternate with light bands (called I bands for isotropic). • The I bands contain only thin (actin) filaments, whereas the A bands contain thick (myosin) filaments. • The myosin and actin filaments overlap in peripheral regions of the A band, whereas a middle region (called the H zone) contains only myosin. Muscle contraction • The basis for understanding muscle contraction is the sliding filament model, first proposed in 1954 both by Andrew Huxley and Ralph Niedergerke and by Hugh Huxley and Jean Hanson • During muscle contraction each sarcomere shortens, bringing the Z discs closer together. • There is no change in the width of the A band, but both the I bands and the H zone almost completely disappear. • These changes are explained by the actin and myosin filaments sliding past one another so that the actin filaments move into the A band and H zone. -

Ciliary Dyneins and Dynein Related Ciliopathies
cells Review Ciliary Dyneins and Dynein Related Ciliopathies Dinu Antony 1,2,3, Han G. Brunner 2,3 and Miriam Schmidts 1,2,3,* 1 Center for Pediatrics and Adolescent Medicine, University Hospital Freiburg, Freiburg University Faculty of Medicine, Mathildenstrasse 1, 79106 Freiburg, Germany; [email protected] 2 Genome Research Division, Human Genetics Department, Radboud University Medical Center, Geert Grooteplein Zuid 10, 6525 KL Nijmegen, The Netherlands; [email protected] 3 Radboud Institute for Molecular Life Sciences (RIMLS), Geert Grooteplein Zuid 10, 6525 KL Nijmegen, The Netherlands * Correspondence: [email protected]; Tel.: +49-761-44391; Fax: +49-761-44710 Abstract: Although ubiquitously present, the relevance of cilia for vertebrate development and health has long been underrated. However, the aberration or dysfunction of ciliary structures or components results in a large heterogeneous group of disorders in mammals, termed ciliopathies. The majority of human ciliopathy cases are caused by malfunction of the ciliary dynein motor activity, powering retrograde intraflagellar transport (enabled by the cytoplasmic dynein-2 complex) or axonemal movement (axonemal dynein complexes). Despite a partially shared evolutionary developmental path and shared ciliary localization, the cytoplasmic dynein-2 and axonemal dynein functions are markedly different: while cytoplasmic dynein-2 complex dysfunction results in an ultra-rare syndromal skeleto-renal phenotype with a high lethality, axonemal dynein dysfunction is associated with a motile cilia dysfunction disorder, primary ciliary dyskinesia (PCD) or Kartagener syndrome, causing recurrent airway infection, degenerative lung disease, laterality defects, and infertility. In this review, we provide an overview of ciliary dynein complex compositions, their functions, clinical disease hallmarks of ciliary dynein disorders, presumed underlying pathomechanisms, and novel Citation: Antony, D.; Brunner, H.G.; developments in the field. -

Kif1b Rab7a Lmna
Title: Charcot-Marie-Tooth Neuropathy Type 2 GeneReview Molecular Genetics: Less Commonly Involved Genes Author: Bird TD Updated: March 2016 KIF1B Gene structure. KIF1B comprises 47 exons and 167.13 kb of DNA. Pathogenic allelic variants. See Table A, Locus Specific and HGMD Normal gene product. Kinesin-like protein KIF1B is involved in axonal transport of synaptic vesicle precursors [Zhao et al 2001]. The kinesin superfamily of proteins is essential for intracellular transport along microtubules. Abnormal gene product. There may be a defect in the transport of synaptic vesicles. RAB7A Gene structure. RAB7A has six exons and 87.9 kb of DNA. Pathogenic allelic variants. See Table A. Normal gene product. Ras-related protein Rab-7a belongs to the RAB family of Ras- related GTPases essential for the regulation of intracellular membrane trafficking. Rab- 7a is involved in transport between late endosomes and lysosomes. RAB-interacting lysosomal protein (RILP) induces the recruitment of dynein-dynactin motors and regulates transport toward the minus-end of microtubules [Verhoeven et al 2003]. Abnormal gene product. Abnormal Rab-7a may cause malfunction of lysosomes and inhibit neurite outgrowth [Spinosa et al 2008, Bucci & Deluca 2012]. LMNA Gene structure. LMNA has 12 exons spread over 24 kb of genomic DNA. Pathogenic allelic variants. The most common pathogenic variant found in individuals with CMT2B1 is p.Arg298Cys, a founder mutation in North Africa [Bouhouche et al 2007, De Sandre-Giovannoli et al 2002]. See also Table A. Table 5. Selected LMNA Variants DNA Nucleotide Protein Amino Acid Class of Variant Allele Reference Sequences Change Change Benign c.1908C>T p.= 1 c.398G>T p.Arg133Leu NM_170707.2 c.892C>T p.Arg298Cys Pathogenic NP_733821.1 c.1411C>T p.Arg471Cys c.1579C>T p.Arg527Cys Note on variant classification: Variants listed in the table have been provided by the author. -
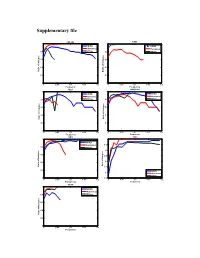
Supplementary File
Supplementary file COL1A2 FZD3 1 1 HPRD HPRD Humannet Humannet 0.8 PPIwu 0.8 PPIwu 0.6 0.6 0.4 0.4 Ratio of of indegree Ratio Ratio of Indegree 0.2 0.2 0 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency RBL1 SMARCA4 1 1 HPRD HPRD Humannet Humannet 0.8 PPIwu 0.8 PPIwu 0.6 0.6 0.4 0.4 Ratio of Indegree Ratio of Indegree Ratio 0.2 0.2 0 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency SRC TP53 1 1 HPRD Humannet 0.95 0.8 PPIwu 0.9 e 0.6 0.85 0.8 0.4 Ratio of Indegree of Ratio Ratio of Indegre 0.75 0.2 HPRD 0.7 Humannet PPIwu 0 0.65 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency VCAN 1 HPRD Humannet 0.8 PPIwu 0.6 0.4 Ratio of Indegree 0.2 0 0 0.05 0.1 0.15 0.2 Frequency Figure S1. Variation of indegree ratio with frequency cutoffs from 0 to 0.2 with a step 0.002 for driver genes common by three fitness cores in COAD. The indegree ratio is set to NULL if total degree of this genes is less than 5. BRAF CASR 1 1 0.8 0.8 0.6 0.6 0.4 0.4 Ratio of Indegree Ratio of Indegree of Ratio HPRD HPRD 0.2 0.2 Humannet Humannet PPIwu PPIwu 0 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency HDAC9 NRAS 1 1 0.8 0.8 0.6 0.6 0.4 0.4 HPRD HPRD Ratio of Indegree Ratio of Indegree 0.2 0.2 Humannet Humannet PPIwu PPIwu 0 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency NF1 CNTNAP2 1 1 0.9 0.8 0.8 0.6 0.7 0.4 HPRD HPRD Ratio of Indegree Ratio of Indegree 0.6 Humannet 0.2 Humannet PPIwu PPIwu 0.5 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency Figure S2.