Metathesis of Functionalized Alkanes: Understanding the Unsolved Problem
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Dramatic Synergy in Copt Nanocatalysts Stabilized by ``Click
Dramatic Synergy in CoPt Nanocatalysts Stabilized by “Click” Dendrimers for Evolution of Hydrogen from Hydrolysis of Ammonia Borane Qi Wang, Fangyu Fu, Sha Yang, Marta Martinez Moro, Maria de los Angeles Ramirez, Sergio Moya, Lionel Salmon, Jaime Ruiz, Didier Astruc To cite this version: Qi Wang, Fangyu Fu, Sha Yang, Marta Martinez Moro, Maria de los Angeles Ramirez, et al.. Dramatic Synergy in CoPt Nanocatalysts Stabilized by “Click” Dendrimers for Evolution of Hydrogen from Hydrolysis of Ammonia Borane. ACS Catalysis, American Chemical Society, 2019, 9 (2), pp.1110- 1119. 10.1021/acscatal.8b04498. hal-02335325 HAL Id: hal-02335325 https://hal.archives-ouvertes.fr/hal-02335325 Submitted on 23 Nov 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. ACS Catalysis This document is confidential and is proprietary to the American Chemical Society and its authors. Do not copy or disclose without written permission. If you have received this item in error, notify the sender and delete all copies. Dramatic Synergy in CoPt Nanocatalysts Stabilized by “Click” -

Richard R. Schrock Department of Chemistry 6-331, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA
MULTIPLE METAL-CARBON BONDS FOR CATALYTIC METATHESIS REACTIONS Nobel Lecture, December 8, 2005 by Richard R. Schrock Department of Chemistry 6-331, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA. It’s my great priviledge to be here today, in a position I never thought pos- sible. I hope the story that I will tell you will give you some idea what I have contributed to the area for which the Nobel Prize in Chemistry was awarded this year. The story begins thirty two years ago in 1973, the year the Nobel Prize was shared by G. Wilkinson and E. O. Fischer. Wilkinson’s Nobel Lecture1 con- cerned the nature of a single bond between a transition metal and a carbon atom in an alkyl group, and emphasized the fact that the metal-carbon bond is not inherently weak. E. O. Fischer in his Nobel Lecture2 summarized the extensive chemistry of transition metal “carbene” complexes3,4 that contain a metal-carbon double bond discovered by him and his group in 1964 (Fig 1).5 He also reported new “carbyne” complexes that contain a metal-carbon triple bond.6 It was clear that metal-carbon single bonds were of great importance in the emerging area of homogeneous catalysis. However, no catalytic reac- tions involving species that contain metal-carbon double or triple bonds were known. When I went to the Central Research Department of E. I. DuPont de Nemours and Company in 1972, transition metal organometallic chemistry and homogeneous catalysis were of great interest as a consequence of their huge potential in organic chemistry and therefore in industry. -

That the Ultimine
THAT THEUS009765271B2ULTIMINE (12 ) United States Patent ( 10 ) Patent No. : US 9 ,765 , 271 B2 Myrick (45 ) Date of Patent: Sep . 19 , 2017 (54 ) NANOPARTICLES, COMPOSITIONS, ( 52 ) U . S . CI. MANUFACTURE AND APPLICATIONS CPC .. .. .. .. CI0L 1 /28 ( 2013 .01 ) ; COIB 33 / 027 (2013 2013 . 01 ) ; C06B 33/ 06 (2013 .01 ) ; C06B 45 /30 (71 ) Applicant: James J . Myrick , Glencoe, IL (US ) ( 2013 .01 ) ; Y10T 428 / 12181 ( 2015 .01 ) ( 58 ) Field of Classification Search ( 72 ) Inventor: James J .Myrick , Glencoe, IL (US ) CPC .. C23C 14 / 0057 ; C01B 33 /037 ( * ) Notice : Subject to any disclaimer , the term of this See application file for complete search history. patent is extended or adjusted under 35 (56 ) References Cited U . S . C . 154 ( b ) by 0 days. U . S . PATENT DOCUMENTS ( 21 ) Appl . No . : 14 / 190 , 082 3 ,008 , 887 A * 11/ 1961 Herglotz . .. C01B 33/ 037 422 / 186 .04 (22 ) Filed : Feb . 25 , 2014 6 ,455 , 455 B1 * 9 /2002 Deller .. .. B82Y 30 /00 423 / 327 . 2 (65 ) Prior Publication Data 2007/ 0158182 A1* 7 / 2007 Takahashi .. .. C23C 14 /0057 204 / 192 . 12 US 2014 /0227548 A1 Aug. 14 , 2014 2008 /0248307 A1 * 10 /2008 Jurbergs .. .. .. .. B82Y 30 /00 Related U . S . Application Data 428 / 405 (63 ) Continuation - in -part of application No . 13 /929 , 771 , * cited by examiner filed on Jun . 27 , 2013 . Primary Examiner — Aileen B Felton (60 ) Provisional application No. 61/ 745 ,810 , filed on Dec . (74 ) Attorney , Agent, or Firm — Husch Blackwell LLP 25 , 2012 , provisional application No . 61/ 664 , 987 , (57 ) ABSTRACT filed on Jun . 27 , 2012 . There are disclosed energetic nanoparticle compositions and materials containing silicon and other energetic elements , (51 ) Int. -

Basic Description for Ground and Air Hazardous
BASIC DESCRIPTION FOR GROUND AND AIR GROUND AND AIR HAZARDOUS MATERIALS SHIPMENTS GROUND SHIPMENTS AIR SHIPMENTS SHIPMENTS HAZARD DOT DOT CLASS OR MAXIMUM EXEMPTION, GROUND EXEMPTION, HAZARDOUS MATERIALS DESCRIPTIONS DIVISION I.D. NUMBER LABEL(S) REQUIRED OR QUANTITY PER SPECIAL SERVICE TO LABEL(S) REQUIRED OR MAXIMUM NET CARGO SPECIAL NON-BULK AND PROPER SHIPPING NAME (Subsidiary if (ALSO MARK PACKING EXEMPTION, SPECIAL PERMIT INNER PERMIT CANADA EXEMPTION, SPECIAL PERMIT QUANTITY PER AIRCRAFT PERMIT SPECIAL EXCEPTIONS PACKAGING (ALSO MARK ON PACKAGE) applicable) ON PACKAGE) GROUP OR EXCEPTION RECEPTACLE OR 173.13 PERMITTED OR EXCEPTION PACKAGE** QUANTITY OR 173.13 PROVISIONS §173.*** §173.*** (1) (2) (3) (4) (5) (6) (7) (8) (9) (10) (11) (12) (13) (14) (15) Accellerene, see p-Nitrosodimethylaniline Accumulators, electric, see Batteries, wet etc Accumulators, pressurized, pneumatic or hydraulic (containing non-flammable gas), see Articles pressurized, pneumatic or hydraulic (containing non-flammable gas) FLAMMABLE FLAMMABLE Acetal 3 UN1088 II LIQUID * YES LIQUID * 5 L 150 202 FLAMMABLE Acetaldehyde 3 UN1089 I LIQUID YES Forbidden None 201 May not be regulated when shipped via UPS Acetaldehyde ammonia 9 UN1841 III ground YES CLASS 9 * 30 kg 30 kg 155 204 FLAMMABLE FLAMMABLE Acetaldehyde oxime 3 UN2332 III LIQUID * YES LIQUID * 25 L 150 203 CORROSIVE, CORROSIVE, Acetic acid, glacial or Acetic acid solution, FLAMMABLE FLAMMABLE A3, A6, with more than 80 percent acid, by mass 8 (3) UN2789 II LIQUID * YES LIQUID * 1 L A7, A10 154 202 Acetic -

1 RICHARD R. SCHROCK [email protected] Field Inorganic And
1 RICHARD R. SCHROCK [email protected] Field Inorganic and Organometallic Chemistry, Catalysis, Polymer Chemistry Current Research Interests Synthetic and mechanistic organo-transition metal chemistry, multiple metal-carbon and nitrogen bonds, homogeneous catalysis, dinitrogen reduction, olefin metathesis, and applications of olefin metathesis to organic and polymer chemistry. Personal Born: January 4, 1945; Berne, Indiana Married: Nancy F. Carlson, 1971; two children Education 1971-1972 Postdoctoral study, Cambridge University, England 1971 Ph.D., Harvard University, Cambridge, Massachusetts 1967 A. B., University of California, Riverside, California Professional Experience 2019-present Professeur Conventionné, ISIS, University of Strasbourg (partial) 2018-present Distinguished Professor and George K. Helmkamp Founder’s Chair of Chemistry, University of California, Riverside (partial) 2018-present Frederick G. Keyes Professor of Chemistry Emeritus, MIT 1989-2018 Frederick G. Keyes Professor of Chemistry, MIT 1980-1989 Professor of Chemistry, MIT 1978-1980 Associate Professor of Chemistry, MIT 1975-1978 Assistant Professor of Chemistry, MIT 1972-1975 Research Chemist, E.I. du Pont de Nemours & Co., CRD Scholarships, Fellowships, Awards, Honorary Degrees 2018 Academician of Honor, Real Academia Europea de Doctores Honorary Professor of Northwest University (China) 2017 James R. Killian Jr. Faculty Achievement Award Honorary Professor of Chengdu University (China) 2015 Humboldt Fellow, Stuttgart Peter Wall Institute Scholar, UBC 2014 Elected -

Imine Metathesis by Silica-Supported Catalysts Using the Methodology of Surface Organometallic Chemistry
Journal of Visualized Experiments www.jove.com Video Article Imine Metathesis by Silica-Supported Catalysts Using the Methodology of Surface Organometallic Chemistry Maha A. Aljuhani1, Jérémie D.A. Pelletier1, Jean-Marie Basset1 1 KAUST Catalysis Center, Division of Physical Sciences and Engineering, King Abdullah University of Science and Technology (KAUST) Correspondence to: Jérémie D.A. Pelletier at [email protected] URL: https://www.jove.com/video/59409 DOI: doi:10.3791/59409 Keywords: Chemistry, Issue 152, surface organometallic chemistry (SOMC), metal-nitrogen fragments, catalysis by design, heterogeneous catalysis, dehydroxylated silica, imine metathesis, well-defined single-site catalysts Date Published: 10/18/2019 Citation: Aljuhani, M.A., Pelletier, J.D., Basset, J.M. Imine Metathesis by Silica-Supported Catalysts Using the Methodology of Surface Organometallic Chemistry. J. Vis. Exp. (152), e59409, doi:10.3791/59409 (2019). Abstract 1 With this protocol, a well-defined singlesite silica-supported heterogeneous catalyst [(≡Si-O-)Hf(=NMe)(η -NMe2)] is designed and prepared according to the methodology developed by surface organometallic chemistry (SOMC). In this framework, catalytic cycles can be determined by isolating crucial intermediates. All air-sensitive materials are handled under inert atmosphere (using gloveboxes or a Schlenk line) or high -5 vacuum lines (HVLs, <10 mbar). The preparation of SiO2-700 (silica dehydroxylated at 700 °C) and subsequent applications (the grafting of complexes and catalytic runs) requires the use of HVLs and double-Schlenk techniques. Several well-known characterization methods are used, such as Fourier-transform infrared spectroscopy (FTIR), elemental microanalysis, solid-state nuclear magnetic resonance spectroscopy (SSNMR), and state-of-the-art dynamic nuclear polarization surface enhanced NMR spectroscopy (DNP-SENS). -
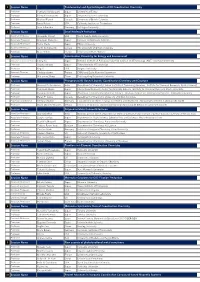
The Session Program
1 Session Name Fundamental and Applied Aspects of N2 Coordination Chemistry Professor Yoshiaki Nishibayashi Japan University of Tokyo Professor Hiroyuki Kawaguchi Japan Tokyo Institute of Technology Professor Michael Fryzuk Canada University of British Columbia Professor Jonas Peters USA California Institute of Technology Professor Sven Schneider Germany Gottingen University 2 Session Name Small Molecule Activation Assistant Professor Alexander Parent USA North Dakota State University Associate Professor Shigeyuki Masaoka Japan Institute for Molecular Science Associate Professor Tohru Wada Japan Rikkyo University Associate Professor Curtis Berlinguette Canada The University of British Columbia Professor Marc Robert France University of Paris Diderot 3 Session Name Coordination Chemistry for Energy and Environment Chief Senior researcher and Professor Qiang Xu Japan National Institute of Advanced Industrial Science and Technology (AIST) and Kobe University Professor Osamu Ishitani Japan Tokyo Institute of Technology Professor Jing Li USA Rutgers University Research Director Christian Serre France CNRS and Ecole Normale Superieure Professor Wai-yeung Wong China The Hong Kong Polytechnic University 4 Session Name Phosphorus(III) Based Ligands: Coordination Chemistry and Catalysis Professor Maravanji S. Balakrishna India Department of Chemistry Indian Institute of Technology Bombay Institute for Chemical Research, Kyoto University Professor Fumiyuki Ozawa Japan International Research Center for Elements Science Institute for Chemical Research, -

Supported Dendrimer-Encapsulated Metal Clusters: Toward Heterogenizing Homogeneous Catalysts † ‡ § † † § † § Rong Ye, , , Aleksandr V
This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. Article pubs.acs.org/accounts Supported Dendrimer-Encapsulated Metal Clusters: Toward Heterogenizing Homogeneous Catalysts † ‡ § † † § † § Rong Ye, , , Aleksandr V. Zhukhovitskiy, Christophe V. Deraedt, , F. Dean Toste,*, , † ‡ § and Gabor A. Somorjai*, , , † ‡ Department of Chemistry, Kavli Energy NanoScience Institute, University of California, Berkeley, Berkeley, California 94720, United States § Chemical Science Division, Lawrence Berkeley National Laboratory, 1 Cyclotron Road, Berkeley, California 94720, United States CONSPECTUS: Recyclable catalysts, especially those that display selective reactivity, are vital for the development of sustainable chemical processes. Among available catalyst platforms, heterogeneous catalysts are particularly well-disposed toward separation from the reaction mixture via filtration methods, which renders them readily recyclable. Furthermore, heterogeneous catalysts offer numer- ous handlessome without homogeneous analoguesfor performance and selectivity optimization. These handles include nano- particle size, pore profile of porous supports, surface ligands and interface with oxide supports, and flow rate through a solid catalyst bed. Despite these available handles, however, conventional heterogeneous catalysts are themselves often structurally heterogeneous compared to homogeneous catalysts, which complicates efforts to optimize and expand the scope of their reactivity and selectivity. Ongoing efforts in our laboratories are aimed to address the above challenge by heterogenizing homogeneous catalysts, which can be defined as the modification of homogeneous catalysts to render them in a separable (solid) phase from the starting materials and products. Specifically, we grow the small nanoclusters in dendrimers, a class of uniform polymers with the connectivity of fractal trees and generally radial symmetry. -

The Remarkable Metal-Catalysed Olefin Metathesis Reaction
Vol 450j8 November 2007jdoi:10.1038/nature06351 REVIEWS The remarkable metal-catalysed olefin metathesis reaction Amir H. Hoveyda1 & Adil R. Zhugralin1 Catalytic olefin metathesis—through which pairs of C5C bonds are reorganized—transforms simple molecules to those that are complex and precious. This class of reactions has noticeably enriched chemical synthesis, which is the art of preparing scarce molecules with highly desirable properties (for example, medicinal agents or polymeric materials). Research in the past two decades has yielded structurally well-defined catalysts for olefin metathesis that are used to synthesize an array of molecules with unprecedented efficiency. Nonetheless, the full potential of olefin metathesis will be realized only when additional catalysts are discovered that are truly practical and afford exceptional selectivity for a significantly broader range of reactions. o appreciate the importance of catalytic olefin metathesis, with relative ease; tri- or tetrasubstituted olefins, on the other hand, we must consider the power of chemical synthesis. The abi- present a challenge owing to higher levels of steric hindrance and lity to prepare molecules is crucial to advances in medicine, complications associated with controlling cis and trans (or E and Z) T biology and materials science1. Chemical synthesis chal- selectivity. Olefin metathesis allows facile access from the easily lenges and expands our understanding of the fundamental principles prepared olefins to those that are cumbersome to access. Efficient of reactivity and selectivity, and gives us the opportunity to examine and stereoselective synthesis of the more substituted olefins is an special molecules that were previously non-existent or available in important and largely unsolved problem in synthesis. -

2020 Emergency Response Guidebook
2020 A guidebook intended for use by first responders A guidebook intended for use by first responders during the initial phase of a transportation incident during the initial phase of a transportation incident involving hazardous materials/dangerous goods involving hazardous materials/dangerous goods EMERGENCY RESPONSE GUIDEBOOK THIS DOCUMENT SHOULD NOT BE USED TO DETERMINE COMPLIANCE WITH THE HAZARDOUS MATERIALS/ DANGEROUS GOODS REGULATIONS OR 2020 TO CREATE WORKER SAFETY DOCUMENTS EMERGENCY RESPONSE FOR SPECIFIC CHEMICALS GUIDEBOOK NOT FOR SALE This document is intended for distribution free of charge to Public Safety Organizations by the US Department of Transportation and Transport Canada. This copy may not be resold by commercial distributors. https://www.phmsa.dot.gov/hazmat https://www.tc.gc.ca/TDG http://www.sct.gob.mx SHIPPING PAPERS (DOCUMENTS) 24-HOUR EMERGENCY RESPONSE TELEPHONE NUMBERS For the purpose of this guidebook, shipping documents and shipping papers are synonymous. CANADA Shipping papers provide vital information regarding the hazardous materials/dangerous goods to 1. CANUTEC initiate protective actions. A consolidated version of the information found on shipping papers may 1-888-CANUTEC (226-8832) or 613-996-6666 * be found as follows: *666 (STAR 666) cellular (in Canada only) • Road – kept in the cab of a motor vehicle • Rail – kept in possession of a crew member UNITED STATES • Aviation – kept in possession of the pilot or aircraft employees • Marine – kept in a holder on the bridge of a vessel 1. CHEMTREC 1-800-424-9300 Information provided: (in the U.S., Canada and the U.S. Virgin Islands) • 4-digit identification number, UN or NA (go to yellow pages) For calls originating elsewhere: 703-527-3887 * • Proper shipping name (go to blue pages) • Hazard class or division number of material 2. -

Catalytic Dehydroaromatization of Alkanes By
CATALYTIC DEHYDROAROMATIZATION OF ALKANES BY PINCER-LIGATED IRIDIUM COMPLEXES By ANDREW MURRAY STEFFENS A dissertation submitted to the Graduate School-New Brunswick Rutgers, The State University of New Jersey In partial fulfillment of the requirements For the degree of Doctor of Philosophy Graduate Program in Chemistry and Chemical Biology Written under the direction of Alan S. Goldman And approved by _______________________________________ _______________________________________ _______________________________________ _______________________________________ New Brunswick, New Jersey January 2016 ABSTRACT OF THE DISSERTATION CATALYTIC DEHYDROAROMATIZATION OF ALKANES BY PINCER-LIGATED IRIDIUM COMPLEXES By ANDREW MURRAY STEFFENS Dissertation Director: Alan S. Goldman Further understanding of the activation of small molecules by metal complexes is an ongoing goal in organometallic chemistry, and expansion on previously reported reactions is of particular interest. This thesis will discuss expansion of transfer dehydrogenation reactions, specifically multiple transfer dehydrogenation reactions of alkanes leading to aromatic products in one pot. The synthesis of the pincer catalyst used for dehydroaromatization reactions is presented along with full characterization of each step. Optimization of dehydroaromatization reactions was performed on a variety of n-alkane starting materials to maximize yield of alkylbenzene products. Kinetics and concentrations for each starting material are presented to highlight the complexity of these reactions. Despite a mechanism that would suggest otherwise, dehydroaromatization reactions surprisingly yield benzene regardless of the starting n-alkane. This interesting appearance of benzene is discussed along with yields of benzene for various n-alkanes. ii Mechanistic studies and DFT calculations were performed to elucidate the mechanism through which benzene is formed, and these studies show that benzene forms though a retro-ene mechanism. -

Distinction with Scholarship The
distinction with scholarship the Tree Issue 22–June 2006 n Other News n 2006 Directory of Members is now available (on request) n List of new members (2005 cycle) elected in 2006 n Letter from the Treasurer to all members – subscriptions n Nomination of new members – process and forms for 2006 cycle n The ‘EUROPEAN REVIEW’ San Souci Palace in Potsdam, see page 28 n This newsletter contains Programmes n Conference and meetings and registration forms for the following announcements events, all of which can be accessed on n Advance notice of events 2006-2007 the website of the Academia (www. n 9th annual conference – acadeuro.org): Toledo 9-2 September 2007 n the 8th Annual meeting of the Academy “Brain, Mind & Matter”, 2-23 September 2006 n workshop (9-20 September): – “Liberty & the Search for Identity” n Workshop (9-20 September): “Grand Challenges of Informatics” Please register for these events NOW by returning the completed forms to the London office as indicated. See inside for reports of n recent workshops and meetings The Klaus Tschira Foundation, see page 18 Academia Europaea’s newsletter–The Tree– From the President The expanding numbers of scholars continent. We are working towards – and we elected 94 in April this year a younger and more balanced and – are all elected after a rigorous peer representative membership that reflects review process. They come from all the the European academic community of disciplines of scholarship and cover the today, and we continue to increase the whole continent and are a testament to number of members from the ‘borders’ the continuing relevance of the Academy of Europe.