Structure and Function of the Bacterial and Fungal Gut Flora of Neotropical Butterflies
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Development and Evaluation of Rrna Targeted in Situ Probes and Phylogenetic Relationships of Freshwater Fungi
Development and evaluation of rRNA targeted in situ probes and phylogenetic relationships of freshwater fungi vorgelegt von Diplom-Biologin Christiane Baschien aus Berlin Von der Fakultät III - Prozesswissenschaften der Technischen Universität Berlin zur Erlangung des akademischen Grades Doktorin der Naturwissenschaften - Dr. rer. nat. - genehmigte Dissertation Promotionsausschuss: Vorsitzender: Prof. Dr. sc. techn. Lutz-Günter Fleischer Berichter: Prof. Dr. rer. nat. Ulrich Szewzyk Berichter: Prof. Dr. rer. nat. Felix Bärlocher Berichter: Dr. habil. Werner Manz Tag der wissenschaftlichen Aussprache: 19.05.2003 Berlin 2003 D83 Table of contents INTRODUCTION ..................................................................................................................................... 1 MATERIAL AND METHODS .................................................................................................................. 8 1. Used organisms ............................................................................................................................. 8 2. Media, culture conditions, maintenance of cultures and harvest procedure.................................. 9 2.1. Culture media........................................................................................................................... 9 2.2. Culture conditions .................................................................................................................. 10 2.3. Maintenance of cultures.........................................................................................................10 -

Genome Diversity and Evolution in the Budding Yeasts (Saccharomycotina)
| YEASTBOOK GENOME ORGANIZATION AND INTEGRITY Genome Diversity and Evolution in the Budding Yeasts (Saccharomycotina) Bernard A. Dujon*,†,1 and Edward J. Louis‡,§ *Department Genomes and Genetics, Institut Pasteur, Centre National de la Recherche Scientifique UMR3525, 75724-CEDEX15 Paris, France, †University Pierre and Marie Curie UFR927, 75005 Paris, France, ‡Centre for Genetic Architecture of Complex Traits, and xDepartment of Genetics, University of Leicester, LE1 7RH, United Kingdom ORCID ID: 0000-0003-1157-3608 (E.J.L.) ABSTRACT Considerable progress in our understanding of yeast genomes and their evolution has been made over the last decade with the sequencing, analysis, and comparisons of numerous species, strains, or isolates of diverse origins. The role played by yeasts in natural environments as well as in artificial manufactures, combined with the importance of some species as model experimental systems sustained this effort. At the same time, their enormous evolutionary diversity (there are yeast species in every subphylum of Dikarya) sparked curiosity but necessitated further efforts to obtain appropriate reference genomes. Today, yeast genomes have been very informative about basic mechanisms of evolution, speciation, hybridization, domestication, as well as about the molecular machineries underlying them. They are also irreplaceable to investigate in detail the complex relationship between genotypes and phenotypes with both theoretical and practical implications. This review examines these questions at two distinct levels offered by the broad evolutionary range of yeasts: inside the best-studied Saccharomyces species complex, and across the entire and diversified subphylum of Saccharomycotina. While obviously revealing evolutionary histories at different scales, data converge to a remarkably coherent picture in which one can estimate the relative importance of intrinsic genome dynamics, including gene birth and loss, vs. -

The Soil Fungal Community of Native Woodland in Andean Patagonian
Forest Ecology and Management 461 (2020) 117955 Contents lists available at ScienceDirect Forest Ecology and Management journal homepage: www.elsevier.com/locate/foreco The soil fungal community of native woodland in Andean Patagonian forest: T A case study considering experimental forest management and seasonal effects ⁎ Ayelen Inés Carrona,b, , Lucas Alejandro Garibaldic, Sebastian Marquezd, Sonia Fontenlaa,b a Laboratorio de Microbiología Aplicada y Biotecnología Vegetal y del Suelo, Centro Regional Universitario Bariloche, Universidad Nacional del Comahue (UNComahue), Argentina b Instituto Andino Patagónico de Tecnologías Biológicas y Geoambientales (IPATEC) UNComahue – Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Argentina c Instituto de Investigaciones en Recursos Naturales, Agroecología y Desarrollo Rural (IRNAD), Sede Andina, Universidad Nacional de Río Negro (UNRN) and CONICET, Argentina d Instituto de Investigación en Biodiversidad y Medio Ambiente (INIBIOMA) UNComahue – Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Argentina ARTICLE INFO ABSTRACT Keywords: Forest management can alter soil fungal communities which are important in the regulation of biogeochemical Soil fungal classification cycles and other ecosystem services. The current challenge of sustainable management is that management be Diversity analysis carried out while preserving the bioecological aspects of ecosystems. Mixed Patagonian woodlands are subject to Shrubland management continuous disturbance (fire, wood -

Saccharomyces Eubayanus, the Missing Link to Lager Beer Yeasts
MICROBE PROFILE Sampaio, Microbiology 2018;164:1069–1071 DOI 10.1099/mic.0.000677 Microbe Profile: Saccharomyces eubayanus, the missing link to lager beer yeasts Jose Paulo Sampaio* Graphical abstract Ecology and phylogeny of Saccharomyces eubayanus. (a) The ecological niche of S. eubayanus in the Southern Hemisphere – Nothofagus spp. (southern beech) and sugar-rich fructifications (stromata) of its fungal biotrophic parasite Cyttaria spp., that can attain the size of golf balls. (b) Schematic representation of the phylogenetic position of S. eubayanus within the genus Saccharomyces based on whole-genome sequences. Occurrence in natural environments (wild) or participation in different human-driven fermentations is highlighted, together with the thermotolerant or cold-tolerant nature of each species and the origins of S. pastorianus, the lager beer hybrid. Abstract Saccharomyces eubayanus was described less than 10 years ago and its discovery settled the long-lasting debate on the origins of the cold-tolerant yeast responsible for lager beer fermentation. The largest share of the genetic diversity of S. eubayanus is located in South America, and strains of this species have not yet been found in Europe. One or more hybridization events between S. eubayanus and S. cerevisiae ale beer strains gave rise to S. pastorianus, the allopolyploid yeasts responsible for lager beer production worldwide. The identification of the missing progenitor of lager yeast opened new avenues for brewing yeast research. It allowed not only the selective breeding of new lager strains, but revealed also a wild yeast with interesting brewing abilities so that a beer solely fermented by S. eubayanus is currently on the market. -

Fungal Planet Description Sheets: 716–784 By: P.W
Fungal Planet description sheets: 716–784 By: P.W. Crous, M.J. Wingfield, T.I. Burgess, G.E.St.J. Hardy, J. Gené, J. Guarro, I.G. Baseia, D. García, L.F.P. Gusmão, C.M. Souza-Motta, R. Thangavel, S. Adamčík, A. Barili, C.W. Barnes, J.D.P. Bezerra, J.J. Bordallo, J.F. Cano-Lira, R.J.V. de Oliveira, E. Ercole, V. Hubka, I. Iturrieta-González, A. Kubátová, M.P. Martín, P.-A. Moreau, A. Morte, M.E. Ordoñez, A. Rodríguez, A.M. Stchigel, A. Vizzini, J. Abdollahzadeh, V.P. Abreu, K. Adamčíková, G.M.R. Albuquerque, A.V. Alexandrova, E. Álvarez Duarte, C. Armstrong-Cho, S. Banniza, R.N. Barbosa, J.-M. Bellanger, J.L. Bezerra, T.S. Cabral, M. Caboň, E. Caicedo, T. Cantillo, A.J. Carnegie, L.T. Carmo, R.F. Castañeda-Ruiz, C.R. Clement, A. Čmoková, L.B. Conceição, R.H.S.F. Cruz, U. Damm, B.D.B. da Silva, G.A. da Silva, R.M.F. da Silva, A.L.C.M. de A. Santiago, L.F. de Oliveira, C.A.F. de Souza, F. Déniel, B. Dima, G. Dong, J. Edwards, C.R. Félix, J. Fournier, T.B. Gibertoni, K. Hosaka, T. Iturriaga, M. Jadan, J.-L. Jany, Ž. Jurjević, M. Kolařík, I. Kušan, M.F. Landell, T.R. Leite Cordeiro, D.X. Lima, M. Loizides, S. Luo, A.R. Machado, H. Madrid, O.M.C. Magalhães, P. Marinho, N. Matočec, A. Mešić, A.N. Miller, O.V. Morozova, R.P. Neves, K. Nonaka, A. Nováková, N.H. -

Evolution of Insect Color Vision: from Spectral Sensitivity to Visual Ecology
EN66CH23_vanderKooi ARjats.cls September 16, 2020 15:11 Annual Review of Entomology Evolution of Insect Color Vision: From Spectral Sensitivity to Visual Ecology Casper J. van der Kooi,1 Doekele G. Stavenga,1 Kentaro Arikawa,2 Gregor Belušic,ˇ 3 and Almut Kelber4 1Faculty of Science and Engineering, University of Groningen, 9700 Groningen, The Netherlands; email: [email protected] 2Department of Evolutionary Studies of Biosystems, SOKENDAI Graduate University for Advanced Studies, Kanagawa 240-0193, Japan 3Department of Biology, Biotechnical Faculty, University of Ljubljana, 1000 Ljubljana, Slovenia; email: [email protected] 4Lund Vision Group, Department of Biology, University of Lund, 22362 Lund, Sweden; email: [email protected] Annu. Rev. Entomol. 2021. 66:23.1–23.28 Keywords The Annual Review of Entomology is online at photoreceptor, compound eye, pigment, visual pigment, behavior, opsin, ento.annualreviews.org anatomy https://doi.org/10.1146/annurev-ento-061720- 071644 Abstract Annu. Rev. Entomol. 2021.66. Downloaded from www.annualreviews.org Copyright © 2021 by Annual Reviews. Color vision is widespread among insects but varies among species, depend- All rights reserved ing on the spectral sensitivities and interplay of the participating photore- Access provided by University of New South Wales on 09/26/20. For personal use only. ceptors. The spectral sensitivity of a photoreceptor is principally determined by the absorption spectrum of the expressed visual pigment, but it can be modified by various optical and electrophysiological factors. For example, screening and filtering pigments, rhabdom waveguide properties, retinal structure, and neural processing all influence the perceived color signal. -

Region of Nymphalidae and Libytheidae (Lepidoptera) from Japan
MORPHOLOGICAL STUDIES ON THE OCCIPITAL REGION OF NYMPHALIDAE AND LIBYTHEIDAE (LEPIDOPTERA) FROM JAPAN Takashi TSUBUKI and Nagao KOYAMA * .h2monji junior High School, 10-33, Kita6tsz{ha 1, Toshima-feu, Toleyo (170), 1opan ** Biological Laboratory, Shinshi2 Udeiversity, Cllada (386), 14pan CONTENTS I. Introduction・--・ny・・・・・・・・・・・・・・・・・-・-・・・・・・-・---・--t・-・---・ny・・・・-・-・・・-・-・・-・・・1 II. Materials and methods・-・・・・・・・・・t・・・・・・t・・・・・・・・・・・・・・`・・・・・・・・・・-・・・-・・・・・・-d・・・・・・・・・・・・2 III. General morphology of the occipital region ・・・・・・}・・・・・・・・・・・・・・・・・・・・・・・・・・・・・4 IV. Occipital structure of each species in Nymphaliclae and Libytheidae ・-ib-・・・・-・・・-・ny・・・・・--・-・・・・・・・-・・-・----・b-・・・----・-・・・-・・-・--・・-6 V. Phylogenical grouping of Nymphalidae based on the occipital structure・t・・・・・・・--・--・・-・-・・・・・・・・・・-・・・・-ny-・・・ny-・--・-----・H"""""".","".."."lg VI. Summary ・・・・-・-・-・・・・・・-・・----・・・-・・・・-・-・-・-----・・-------・-・----・23 VII. Literatures cited ・・・--・・・・・・・・・`・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・--・-・・-・・・・・・tt・・・・・・23 I. INTRODUCTION A considerable number of studies have been carried out on the occipital morphology of moths since YAGI and KoYAMA's report of 1963 (KoYAMA and MlyATA, 1969, 1970, 1975;MIYATA, 1971, 1972, 1973, 1974, 1975;MIyATA and KOYAMA, 1971, 1972, 1976). In the butterfiies, however, few papers were available concerning the occipital region, on which EHRucH (1958, a b), YAGI and KoyAMA (1963) and KoyAMA and OGAwA (1972) briefly described. Then, tried to study the occipital region of butterflies, the authors observed it preliminarily (TsuBuKI et aL 1975). The present paper deals with the occipital structure of Nymphalidae and Libytheidae from Japan with reference to its bearing on systematics. Before going further, the authors wish to express their gratitude to Prof. Dr. H. SAwADA and Prof. Dr. N. GOKAN, Tokyo University of Agriculture, for their advice and assistance. Thanks are also due to Mr. N. K6DA who gave valuable materials for this work, and to Mr. Y. YAGucm and the members of JEOL Ltd. -
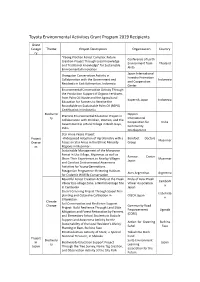
List of Previous Grant Projects
Toyota Environmental Activities Grant Program 2019 Recipients Grant Catego Theme Project Description Organization Country ry "Kaeng Krachan Forest Complex: Future Conference of Earth Creation Project Through Local Knowledge Environment from Thailand and Traditional Knowledge" for Sustainable Akita Environmental Innovation Japan International Orangutan Conservation Activity in Forestry Promotion Collaboration with the Government and Indonesia and Cooperation Residents in East Kalimantan, Indonesia Center Environmental Conservation Activity Through the Production Support of Organic Fertilizers from Palm Oil Waste and the Agricultural Kopernik Japan Indonesia Education for Farmers to Receive the Roundtable on Sustainable Palm Oil (RSPO) Certification in Indonesia Biodiversi Nippon Practical Environmental Education Project in ty International Collaboration with Children, Women, and the Cooperation for India Government in a Rural Village in Bodh Gaya, Community India Development Star Anise Peace Project Project -Widespread Adoption of Agroforestry with a Barefoot Doctors Myanmar Overse Focus on Star Anise in the Ethnic Minority Group as Regions in Myanmar- Sustainable Management of the Mangrove Forest in Uto Village, Myanmar, as well as Ramsar Center Share Their Experiences to Nearby Villages Myanmar Japan and Conduct Environmental Awareness Activities for Young Generations Patagonian Programme: Restoring Habitats Aves Argentinas Argentina for Endemic Wildlife Conservation Beautiful Forest Creation Activity at the Preah Pride of Asia: Preah -

Epiphytic Seed Microbiomes of Wheat, Canola, and Lentil
EPIPHYTIC SEED MICROBIOMES OF WHEAT, CANOLA, AND LENTIL A Thesis Submitted to the College of Graduate and Postdoctoral Studies In Partial Fulfillment of the Requirements For the Degree of Doctor of Philosophy In the Department of Food and Bioproduct Sciences University of Saskatchewan Saskatoon By Zayda Piedad Morales Moreira © Copyright Zayda Piedad Morales Moreira, June, 2021. All rights reserved. Unless otherwise noted, copyright of the material in this thesis belongs to the author PERMISSION TO USE In presenting this thesis in partial fulfilment of the requirements for a Postgraduate degree from the University of Saskatchewan, I agree that the Libraries of this University may make it freely available for inspection. I further agree that permission for copying of this thesis in any manner, in whole or in part, for scholarly purposes may be granted by the professor or professors who supervised my thesis work or, in their absence, by the Head of the Department or the Dean of the College in which my thesis work was done. It is understood that any copying, publication, or use of this thesis or parts thereof for financial gain shall not be allowed without my written permission. It is also understood that due recognition shall be given to me and to the University of Saskatchewan in any scholarly use which may be made of any material in my thesis. Requests for permission to copy or to make other use of material in this thesis in whole or part should be addressed to: Head of the Department of Food and Bioproduct Sciences University of Saskatchewan 51 Campus Drive University of Saskatchewan Saskatoon, Saskatchewan, S7N 5A8 Canada OR Dean of the College of Graduate and Postdoctoral Studies University of Saskatchewan 107 Administration Place Saskatoon, Saskatchewan S7N 5A2 Canada i ABSTRACT Microorganisms are found colonizing all plant organs including seeds. -

Genomic Diversity Landscape of the Honey Bee Gut Microbiota
ARTICLE https://doi.org/10.1038/s41467-019-08303-0 OPEN Genomic diversity landscape of the honey bee gut microbiota Kirsten M. Ellegaard 1 & Philipp Engel 1 The structure and distribution of genomic diversity in natural microbial communities is largely unexplored. Here, we used shotgun metagenomics to assess the diversity of the honey bee gut microbiota, a community consisting of few bacterial phylotypes. Our results show that 1234567890():,; most phylotypes are composed of sequence-discrete populations, which co-exist in individual bees and show age-specific abundance profiles. In contrast, strains present within these sequence-discrete populations were found to segregate into individual bees. Consequently, despite a conserved phylotype composition, each honey bee harbors a distinct community at the functional level. While ecological differentiation seems to facilitate coexistence at higher taxonomic levels, our findings suggest that, at the level of strains, priority effects during community assembly result in individualized profiles, despite the social lifestyle of the host. Our study underscores the need to move beyond phylotype-level characterizations to understand the function of this community, and illustrates its potential for strain-level analysis. 1 Department of Fundamental Microbiology, University of Lausanne, 1015 Lausanne, Switzerland. Correspondence and requests for materials should be addressed to K.M.E. (email: [email protected]) or to P.E. (email: [email protected]) NATURE COMMUNICATIONS | (2019) 10:446 | https://doi.org/10.1038/s41467-019-08303-0 | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-019-08303-0 ost bacteria live in genetically diverse and highly com- same species name22. -

Crittendenia Gen. Nov., a New Lichenicolous Lineage in the Agaricostilbomycetes (Pucciniomycotina), and a Review of the Biology
The Lichenologist (2021), 53, 103–116 doi:10.1017/S002428292000033X Standard Paper Crittendenia gen. nov., a new lichenicolous lineage in the Agaricostilbomycetes (Pucciniomycotina), and a review of the biology, phylogeny and classification of lichenicolous heterobasidiomycetes Ana M. Millanes1, Paul Diederich2, Martin Westberg3 and Mats Wedin4 1Departamento de Biología y Geología, Física y Química Inorgánica, Universidad Rey Juan Carlos, E-28933 Móstoles, Spain; 2Musée national d’histoire naturelle, 25 rue Munster, L-2160 Luxembourg; 3Museum of Evolution, Norbyvägen 16, SE-75236 Uppsala, Sweden and 4Department of Botany, Swedish Museum of Natural History, P.O. Box 50007, SE-10405 Stockholm, Sweden Abstract The lichenicolous ‘heterobasidiomycetes’ belong in the Tremellomycetes (Agaricomycotina) and in the Pucciniomycotina. In this paper, we provide an introduction and review of these lichenicolous taxa, focusing on recent studies and novelties of their classification, phylogeny and evolution. Lichen-inhabiting fungi in the Pucciniomycotina are represented by only a small number of species included in the genera Chionosphaera, Cyphobasidium and Lichenozyma. The phylogenetic position of the lichenicolous representatives of Chionosphaera has, however, never been investigated by molecular methods. Phylogenetic analyses using the nuclear SSU, ITS, and LSU ribosomal DNA mar- kers reveal that the lichenicolous members of Chionosphaera form a monophyletic group in the Pucciniomycotina, distinct from Chionosphaera and outside the Chionosphaeraceae. The new genus Crittendenia is described to accommodate these lichen-inhabiting spe- cies. Crittendenia is characterized by minute synnemata-like basidiomata, the presence of clamp connections and aseptate tubular basidia from which 4–7 spores discharge passively, often in groups. Crittendenia, Cyphobasidium and Lichenozyma are the only lichenicolous lineages known so far in the Pucciniomycotina, whereas Chionosphaera does not include any lichenicolous taxa. -

Photonic Crystal Structure of Wing Scales in Sasakia Charonda Butterflies
Materials Transactions, Vol. 51, No. 2 (2010) pp. 202 to 208 Special Issue on Development and Fabrication of Advanced Materials Assisted by Nanotechnology and Microanalysis #2010 The Japan Institute of Metals Photonic Crystal Structure of Wing Scales in Sasakia Charonda Butterflies Jirˇina Mateˇjkova´-Plsˇkova´1, Dalibor Jancˇik1, Miroslav Masˇla´nˇ1, Satoshi Shiojiri2 and Makoto Shiojiri3;* 1Centre for Nanomaterial Research, Faculty of Science, Palacky University in Olomouc, Slechtitelu 11, 783 71 Olomouc, Czech Republic 2Matsubara Junior High-school, Kyoto 604-8812, Japan 3Professor Emeritus of Kyoto Institute of Technology, 1-297 Wakiyama, Kyoto 618-0091, Japan The hindwings of the male Sasakia charonda charonda butterflies comprise iridescent purple-blue areas, iridescent white-pearl areas, yellow spots and red spots as well as brown background. We have examined the microstructure of their scales by scanning electron microscopy, for applying their photonic crystal structures to fine light manipulators such as reflection elements in laser diodes. The scales in the yellow spots, red spots and brown background have almost the same structure, which is an optical diffraction grating made of ridges with two cuticle layers. Their difference comes from the contained pigments. The scales in the iridescent purple-blue and white-pearl are also the same in structure. They have seven tilted cuticle layers lapped on the ridges, which also constitute a grating. The widths of the ridge and groove in the grating are different between scales of the two kinds. It is shown that the vivid iridescence is mainly attributed to multiple interferences caused between rays reflected from the seven cuticle layers with air gaps.