From Metagenomics to Bioproducts
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The 2014 Golden Gate National Parks Bioblitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event
National Park Service U.S. Department of the Interior Natural Resource Stewardship and Science The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 ON THIS PAGE Photograph of BioBlitz participants conducting data entry into iNaturalist. Photograph courtesy of the National Park Service. ON THE COVER Photograph of BioBlitz participants collecting aquatic species data in the Presidio of San Francisco. Photograph courtesy of National Park Service. The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 Elizabeth Edson1, Michelle O’Herron1, Alison Forrestel2, Daniel George3 1Golden Gate Parks Conservancy Building 201 Fort Mason San Francisco, CA 94129 2National Park Service. Golden Gate National Recreation Area Fort Cronkhite, Bldg. 1061 Sausalito, CA 94965 3National Park Service. San Francisco Bay Area Network Inventory & Monitoring Program Manager Fort Cronkhite, Bldg. 1063 Sausalito, CA 94965 March 2016 U.S. Department of the Interior National Park Service Natural Resource Stewardship and Science Fort Collins, Colorado The National Park Service, Natural Resource Stewardship and Science office in Fort Collins, Colorado, publishes a range of reports that address natural resource topics. These reports are of interest and applicability to a broad audience in the National Park Service and others in natural resource management, including scientists, conservation and environmental constituencies, and the public. The Natural Resource Report Series is used to disseminate comprehensive information and analysis about natural resources and related topics concerning lands managed by the National Park Service. -

Diversity of Understudied Archaeal and Bacterial Populations of Yellowstone National Park: from Genes to Genomes Daniel Colman
University of New Mexico UNM Digital Repository Biology ETDs Electronic Theses and Dissertations 7-1-2015 Diversity of understudied archaeal and bacterial populations of Yellowstone National Park: from genes to genomes Daniel Colman Follow this and additional works at: https://digitalrepository.unm.edu/biol_etds Recommended Citation Colman, Daniel. "Diversity of understudied archaeal and bacterial populations of Yellowstone National Park: from genes to genomes." (2015). https://digitalrepository.unm.edu/biol_etds/18 This Dissertation is brought to you for free and open access by the Electronic Theses and Dissertations at UNM Digital Repository. It has been accepted for inclusion in Biology ETDs by an authorized administrator of UNM Digital Repository. For more information, please contact [email protected]. Daniel Robert Colman Candidate Biology Department This dissertation is approved, and it is acceptable in quality and form for publication: Approved by the Dissertation Committee: Cristina Takacs-Vesbach , Chairperson Robert Sinsabaugh Laura Crossey Diana Northup i Diversity of understudied archaeal and bacterial populations from Yellowstone National Park: from genes to genomes by Daniel Robert Colman B.S. Biology, University of New Mexico, 2009 DISSERTATION Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Biology The University of New Mexico Albuquerque, New Mexico July 2015 ii DEDICATION I would like to dedicate this dissertation to my late grandfather, Kenneth Leo Colman, associate professor of Animal Science in the Wool laboratory at Montana State University, who even very near the end of his earthly tenure, thought it pertinent to quiz my knowledge of oxidized nitrogen compounds. He was a man of great curiosity about the natural world, and to whom I owe an acknowledgement for his legacy of intellectual (and actual) wanderlust. -

Phylogenetics of Archaeal Lipids Amy Kelly 9/27/2006 Outline
Phylogenetics of Archaeal Lipids Amy Kelly 9/27/2006 Outline • Phlogenetics of Archaea • Phlogenetics of archaeal lipids • Papers Phyla • Two? main phyla – Euryarchaeota • Methanogens • Extreme halophiles • Extreme thermophiles • Sulfate-reducing – Crenarchaeota • Extreme thermophiles – Korarchaeota? • Hyperthermophiles • indicated only by environmental DNA sequences – Nanoarchaeum? • N. equitans a fast evolving euryarchaeal lineage, not novel, early diverging archaeal phylum – Ancient archael group? • In deepest brances of Crenarchaea? Euryarchaea? Archaeal Lipids • Methanogens – Di- and tetra-ethers of glycerol and isoprenoid alcohols – Core mostly archaeol or caldarchaeol – Core sometimes sn-2- or Images removed due to sn-3-hydroxyarchaeol or copyright considerations. macrocyclic archaeol –PMI • Halophiles – Similar to methanogens – Exclusively synthesize bacterioruberin • Marine Crenarchaea Depositional Archaeal Lipids Biological Origin Environment Crocetane methanotrophs? methane seeps? methanogens, PMI (2,6,10,15,19-pentamethylicosane) methanotrophs hypersaline, anoxic Squalane hypersaline? C31-C40 head-to-head isoprenoids Smit & Mushegian • “Lost” enzymes of MVA pathway must exist – Phosphomevalonate kinase (PMK) – Diphosphomevalonate decarboxylase – Isopentenyl diphosphate isomerase (IPPI) Kaneda et al. 2001 Rohdich et al. 2001 Boucher et al. • Isoprenoid biosynthesis of archaea evolved through a combination of processes – Co-option of ancestral enzymes – Modification of enzymatic specificity – Orthologous and non-orthologous gene -

Supporting Information
Supporting Information Lozupone et al. 10.1073/pnas.0807339105 SI Methods nococcus, and Eubacterium grouped with members of other Determining the Environmental Distribution of Sequenced Genomes. named genera with high bootstrap support (Fig. 1A). One To obtain information on the lifestyle of the isolate and its reported member of the Bacteroidetes (Bacteroides capillosus) source, we looked at descriptive information from NCBI grouped firmly within the Firmicutes. This taxonomic error was (www.ncbi.nlm.nih.gov/genomes/lproks.cgi) and other related not surprising because gut isolates have often been classified as publications. We also determined which 16S rRNA-based envi- Bacteroides based on an obligate anaerobe, Gram-negative, ronmental surveys of microbial assemblages deposited near- nonsporulating phenotype alone (6, 7). A more recent 16S identical sequences in GenBank. We first downloaded the gbenv rRNA-based analysis of the genus Clostridium defined phylo- files from the NCBI ftp site on December 31, 2007, and used genetically related clusters (4, 5), and these designations were them to create a BLAST database. These files contain GenBank supported in our phylogenetic analysis of the Clostridium species in the HGMI pipeline. We thus designated these Clostridium records for the ENV database, a component of the nonredun- species, along with the species from other named genera that dant nucleotide database (nt) where 16S rRNA environmental cluster with them in bootstrap supported nodes, as being within survey data are deposited. GenBank records for hits with Ͼ98% these clusters. sequence identity over 400 bp to the 16S rRNA sequence of each of the 67 genomes were parsed to get a list of study titles Annotation of GTs and GHs. -

Archaeology of Eukaryotic DNA Replication
Downloaded from http://cshperspectives.cshlp.org/ on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press Archaeology of Eukaryotic DNA Replication Kira S. Makarova and Eugene V. Koonin National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland 20894 Correspondence: [email protected] Recent advances in the characterization of the archaeal DNA replication system together with comparative genomic analysis have led to the identification of several previously un- characterized archaeal proteins involved in replication and currently reveal a nearly com- plete correspondence between the components of the archaeal and eukaryotic replication machineries. It can be inferred that the archaeal ancestor of eukaryotes and even the last common ancestor of all extant archaea possessed replication machineries that were compa- rable in complexity to the eukaryotic replication system. The eukaryotic replication system encompasses multiple paralogs of ancestral components such that heteromeric complexes in eukaryotes replace archaeal homomeric complexes, apparently along with subfunctionali- zation of the eukaryotic complex subunits. In the archaea, parallel, lineage-specific dupli- cations of many genes encoding replication machinery components are detectable as well; most of these archaeal paralogs remain to be functionally characterized. The archaeal rep- lication system shows remarkable plasticity whereby even some essential components such as DNA polymerase and single-stranded DNA-binding protein are displaced by unrelated proteins with analogous activities in some lineages. ouble-stranded DNA is the molecule that Okazaki fragments (Kornberg and Baker 2005; Dcarries genetic information in all cellular Barry and Bell 2006; Hamdan and Richardson life-forms; thus, replication of this genetic ma- 2009; Hamdan and van Oijen 2010). -
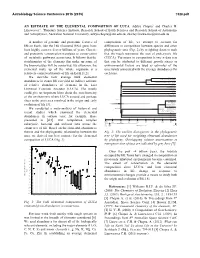
An Estimate of the Elemental Composition of Luca
Astrobiology Science Conference 2015 (2015) 7328.pdf AN ESTIMATE OF THE ELEMENTAL COMPOSITION OF LUCA. Aditya Chopra1 and Charles H. Lineweaver1, 1Planetary Science Institute, Research School of Earth Sciences and Research School of Astronomy and Astrophysics, Australian National University, [email protected], [email protected] A number of genomic and proteomic features of composition of life, we attempt to account for life on Earth, like the 16S ribosomal RNA gene, have differences in composition between species and other been highly conserved over billions of years. Genetic phylogenetic taxa (Fig. 2) by weighting datasets such and proteomic conservation translates to conservation that the result represents the root of prokaryotic life of metabolic pathways across taxa. It follows that the (LUCA). Variations in composition between data sets stoichiometry of the elements that make up some of that can be attributed to different growth stages or the biomolecules will be conserved. By extension, the environmental factors are used as estimates of the elemental make up of the whole organism is a uncertainty associated with the average abundances for relatively conserved feature of life on Earth [1,2]. each taxa. We describe how average bulk elemental Euryarchaeota 1a Methanococci, Methanobacteria, Methanopyri 7 Euryarchaeota 1b abundances in extant life can yield an indirect estimate 6 Thermoplasmata, Methanomicrobia, Halobacteria, Archaeoglobi Euryarchaeota 2 4 Thermococci of relative abundances of elements in the Last Crenarchaeota Sulfolobus, Thermoproteus ? Thaumarchaeota Universal Common Ancestor (LUCA). The results Cenarchaeum ? Korarchaeota ARMAN could give us important hints about the stoichiometry ? 2 Archaeal Richmond Mine Acidophilic Nanoorganisms Nanoarchaeota of the environment where LUCA existed and perhaps Archaea Terrabacteria clues to the processes involved in the origin and early Actinobacteria, Deinococcus-Thermus, 1 Cyanobacteria, Life Chloroflexi, 9 evolution of life [3]. -

Brian W. Waters
INVESTIGATION OF 2-OXOACID OXIDOREDUCTASES IN METHANOCOCCUS MARIPALUDIS AND LARGE-SCALE GROWTH OF M. MARIPALUDIS by BRIAN W. WATERS (Under the direction of Dr. William B. Whitman) ABSTRACT With the emergence of Methanococcus maripaludis as a genetic model for methanogens, it becomes imperative to devise inexpensive yet effective ways to grow large numbers of cells for protein studies. Chapter 2 details methods that were used to cut costs of growing M. maripaludis in large scale. Also, large scale growth under different conditions was explored in order to find the conditions that yield the most cells. Chapter 3 details the phylogenetic analysis of 2-oxoacid oxidoreductase (OR) homologs from M. maripaludis. ORs are enzymes that catalyze the oxidative decarboxylation of 2-oxoacids to their acyl-CoA derivatives in many prokaryotes. Also in chapter 3, a specific OR homolog in M. maripaludis, an indolepyruvate oxidoreductase (IOR), was mutagenized, and the mutant was characterized. PCR and Southern hybridization analysis showed gene replacement. A no-growth phenotype on media containing aromatic amino acid derivatives was found. INDEX WORDS: Methanococcus maripaludis, fermentor, 2-oxoacid oxidoreductase, indolepyruvate oxidoreductase INVESTIGATION OF 2-OXOACID OXIDOREDUCTASES IN METHANOCOCCUS MARIPALUDIS AND LARGE-SCALE GROWTH OF M. MARIPALUDIS by BRIAN W. WATERS B.S., The University of Georgia,1999 A Thesis Submitted to the Graduate Faculty of The University of Georgia in Partial Fulfillment of the Requirements for the Degree MASTER OF SCIENCE ATHENS, GEORGIA 2002 © 2002 Brian W. Waters All Rights Reserved INVESTIGATION OF 2-OXOACID OXIDOREDUCTASES IN METHANOCOCCUS MARIPALUDIS AND LARGE-SCALE GROWTH OF M. MARIPALUDIS by BRIAN W. -

An Integrated Study on Microbial Community in Anaerobic Digestion Systems
An Integrated Study on Microbial Community in Anaerobic Digestion Systems DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Yueh-Fen Li Graduate Program in Environmental Science The Ohio State University 2013 Dissertation Committee: Dr. Zhongtang Yu, Advisor Dr. Brian Ahmer Dr. Richard Dick Dr. Olli Tuovinen Copyrighted by Yueh-Fen Li 2013 Abstract Anaerobic digestion (AD) is an attractive microbiological technology for both waste treatment and energy production. Microorganisms are the driving force for the whole transformation process in anaerobic digesters. However, the microbial community underpinning the AD process remains poorly understood, especially with respect to community composition and dynamics in response to variations in feedstocks and operations. The overall objective was to better understand the microbiology driving anaerobic digestion processes by systematically investigating the diversity, composition and succession of microbial communities, both bacterial and archaeal, in anaerobic digesters of different designs, fed different feedstocks, and operated under different conditions. The first two studies focused on propionate-degrading bacteria with an emphasis on syntrophic propionate-oxidizing bacteria. Propionate is one of the most important intermediates and has great influence on AD stability in AD systems because it is inhibitory to methanogens and it can only be metabolized through syntrophic propionate- oxidizing acetogenesis under methanogenic conditions. In the first study (chapter 3), primers specific to the propionate-CoA transferase gene (pct) were designed and used to construct clone libraries, which were sequenced and analyzed to investigate the diversity and distribution of propionate-utilizing bacteria present in the granular and the liquid portions of samples collected from four digesters of different designs, fed different ii feedstocks, and operated at different temperatures. -

Universidad Politécnica De Madrid Escuela Técnica Superior De Ingeniería Agronómica Alimentaria Y De Biosistemas Departamento De Biotecnología-Biología Vegetal
UNIVERSIDAD POLITÉCNICA DE MADRID ESCUELA TÉCNICA SUPERIOR DE INGENIERÍA AGRONÓMICA ALIMENTARIA Y DE BIOSISTEMAS DEPARTAMENTO DE BIOTECNOLOGÍA-BIOLOGÍA VEGETAL TESIS DOCTORAL Caracterización de una nueva proteína hipertermófila NifB y su papel en el mecanismo de síntesis de NifB-co, precursor del grupo metálico de FeMo-co Author: Alessandro Scandurra Director: Prof. Luis Manuel Rubio Herrero Co-director: Dr. Simon Jean Marius Arragain i ii UNIVERSIDAD POLITÉCNICA DE MADRID ESCUELA TÉCNICA SUPERIOR DE INGENIERÍA AGRONÓMICA ALIMENTARIA Y DE BIOSISTEMAS DEPARTAMENTO DE BIOTECNOLOGÍA-BIOLOGÍA VEGETAL Memoria presentada por D. Alessandro Scandurra para optar al grado de Doctor Director Dr. Luis Manuel Rubio Herrero Profesor Titular UPM Madrid, 2017 © This copy oF the thesis has been supplied on condition that anyone who consults it is understood to recognize that its copyright rest with the author and that no quotation From the thesis, or any inFormation derived therefrom may be published without the author’s prior, written consent. I II AcKnowledgments As each journey, also this one has an end (Thanks goodness). It was a long trip, with several changes oF direction. Like For many other people, the PhD adventure was not easy, but I have to admit that I was lucky enough to meet really unique people, that became special Friendships. First oF all, I want to thank my Family, especially my mother and my Father, who always push me to go Forward in my life and to Follow my dreams, ready to pay the sacrifice oF the distance. This expirience has helped me to understand how easy is to be a son and how challenging is to be a parent. -

Methanogens Diversity During Anaerobic Sewage Sludge Stabilization and the Effect of Temperature
processes Article Methanogens Diversity during Anaerobic Sewage Sludge Stabilization and the Effect of Temperature Tomáš Vítˇez 1,2, David Novák 3, Jan Lochman 3,* and Monika Vítˇezová 1,* 1 Department of Experimental Biology, Faculty of Science, Masaryk University, 62500 Brno, Czech Republic; [email protected] 2 Department of Agricultural, Food and Environmental Engineering, Faculty of AgriSciences, Mendel University, 61300 Brno, Czech Republic 3 Department of Biochemistry, Faculty of Science, Masaryk University, 62500 Brno, Czech Republic; [email protected] * Correspondence: [email protected] (J.L.); [email protected] (M.V.); Tel.: +420-549-495-602 (J.L.); Tel.: +420-549-497-177 (M.V.) Received: 29 June 2020; Accepted: 10 July 2020; Published: 12 July 2020 Abstract: Anaerobic sludge stabilization is a commonly used technology. Most fermenters are operated at a mesophilic temperature regime. Modern trends in waste management aim to minimize waste generation. One of the strategies can be achieved by anaerobically stabilizing the sludge by raising the temperature. Higher temperatures will allow faster decomposition of organic matter, shortening the retention time, and increasing biogas production. This work is focused on the description of changes in the community of methanogenic microorganisms at different temperatures during the sludge stabilization. At higher temperatures, biogas contained a higher percentage of methane, however, there was an undesirable accumulation of ammonia in the fermenter. Representatives of the hydrogenotrophic genus Methanoliea were described at all temperatures tested. At temperatures up to 50 ◦C, a significant proportion of methanogens were also formed by acetoclastic representatives of Methanosaeta sp. and acetoclastic representatives of the order Methanosarcinales. -

Article Mode, Which Adds to a Better and Living Conditions of Microorganisms in the Atmosphere Understanding of the Overall Atmospheric Microbiome
Biogeosciences, 15, 4205–4214, 2018 https://doi.org/10.5194/bg-15-4205-2018 © Author(s) 2018. This work is distributed under the Creative Commons Attribution 4.0 License. Community composition and seasonal changes of archaea in coarse and fine air particulate matter Jörn Wehking1,2, Daniel A. Pickersgill1,2, Robert M. Bowers3,4, David Teschner1,2, Ulrich Pöschl2, Janine Fröhlich-Nowoisky2, and Viviane R. Després1,2 1Institute of Molecular Physiology, Johannes Gutenberg University, Johannes-von-Müller-Weg 6, 55128 Mainz, Germany 2Max Planck Institute for Chemistry, P.O. Box 3060, 55020 Mainz, Germany 3DOE Joint Genome Institute, Walnut Creek, CA, USA 4University of Colorado, Ecology and Evolutionary Biology, Boulder, CO, USA Correspondence: Viviane R. Després ([email protected]) and Jörn Wehking ([email protected]) Received: 1 December 2017 – Discussion started: 14 December 2017 Revised: 11 June 2018 – Accepted: 15 June 2018 – Published: 11 July 2018 Abstract. Archaea are ubiquitous in terrestrial and marine narchaeaceae, Nitrososphaeraceae, Methanosarcinales, Ther- environments and play an important role in biogeochemical moplasmata, and the genus Nitrosopumilus as the dominating cycles. Although air acts as the primary medium for their taxa. dispersal among different habitats, their diversity and abun- The seasonal dynamics of methanogenic Euryarchaeota dance is not well characterized. The main reason for this point to anthropogenic activities, such as fertilization of agri- lack of insight is that archaea are difficult to culture, seem cultural fields with biogas substrates or manure, as sources of to be low in number in the atmosphere, and have so far been airborne archaea. This study gains a deeper insight into the difficult to detect even with molecular genetic approaches. -

(Gid ) Genes Coding for Putative Trna:M5u-54 Methyltransferases in 355 Bacterial and Archaeal Complete Genomes
Table S1. Taxonomic distribution of the trmA and trmFO (gid ) genes coding for putative tRNA:m5U-54 methyltransferases in 355 bacterial and archaeal complete genomes. Asterisks indicate the presence and the number of putative genes found. Genomes Taxonomic position TrmA Gid Archaea Crenarchaea Aeropyrum pernix_K1 Crenarchaeota; Thermoprotei; Desulfurococcales; Desulfurococcaceae Cenarchaeum symbiosum Crenarchaeota; Thermoprotei; Cenarchaeales; Cenarchaeaceae Pyrobaculum aerophilum_str_IM2 Crenarchaeota; Thermoprotei; Thermoproteales; Thermoproteaceae Sulfolobus acidocaldarius_DSM_639 Crenarchaeota; Thermoprotei; Sulfolobales; Sulfolobaceae Sulfolobus solfataricus Crenarchaeota; Thermoprotei; Sulfolobales; Sulfolobaceae Sulfolobus tokodaii Crenarchaeota; Thermoprotei; Sulfolobales; Sulfolobaceae Euryarchaea Archaeoglobus fulgidus Euryarchaeota; Archaeoglobi; Archaeoglobales; Archaeoglobaceae Haloarcula marismortui_ATCC_43049 Euryarchaeota; Halobacteria; Halobacteriales; Halobacteriaceae; Haloarcula Halobacterium sp Euryarchaeota; Halobacteria; Halobacteriales; Halobacteriaceae; Haloarcula Haloquadratum walsbyi Euryarchaeota; Halobacteria; Halobacteriales; Halobacteriaceae; Haloquadra Methanobacterium thermoautotrophicum Euryarchaeota; Methanobacteria; Methanobacteriales; Methanobacteriaceae Methanococcoides burtonii_DSM_6242 Euryarchaeota; Methanomicrobia; Methanosarcinales; Methanosarcinaceae Methanococcus jannaschii Euryarchaeota; Methanococci; Methanococcales; Methanococcaceae Methanococcus maripaludis_S2 Euryarchaeota; Methanococci;