Experiment #3: Asymmetric Synthesis – Use of a Chiral Manganese Catalyst for the Enantioselective Epoxidation of Alkenes†
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chiral Separation of Rac-Propylene Oxide on Penicillamine Coated Gold Nps
nanomaterials Article Chiral Separation of rac-Propylene Oxide on Penicillamine Coated Gold NPs Nisha Shukla 1,2, Zachary Blonder 2 and Andrew J. Gellman 2,3,* 1 Institute of Complex Engineered Systems, Carnegie Mellon University, Pittsburgh, PA 15213, USA; [email protected] 2 Department of Chemical Engineering, Carnegie Mellon University, Pittsburgh, PA 15213, USA; [email protected] 3 W.E. Scott Institute for Energy Innovation, Carnegie Mellon University, Pittsburgh, PA 15213, USA * Correspondence: [email protected] Received: 16 July 2020; Accepted: 20 August 2020; Published: 30 August 2020 Abstract: The surfaces of chemically synthesized spherical gold NPs (Au-NPs) have been modified using chiral L- or D-penicillamine (Pen) in order to impart enantioselective adsorption properties. These chiral Au-NPs have been used to demonstrate enantioselective adsorption of racemic propylene oxide (PO) from aqueous solution. In the past we have studied enantioselective adsorption of racemic PO on L- or D-cysteine (Cys)-coated Au-NPs. This prior work suggested that adsorption of PO on Cys-coated Au-NPs equilibrates within an hour. In this work, we have studied the effect of time on the enantioselective adsorption of racemic PO from solution onto chiral Pen/Au-NPs. Enantioselective adsorption of PO on chiral Pen/Au-NPs is time-dependent but reaches a steady state after ~18 h at room temperature. More importantly, L- or D-Pen/Au-NPs are shown to adsorb R- or S-PO enantiospecifically and to separate the two PO enantiomers from racemic mixtures of RS-PO. Keywords: chiral; nanoparticles; enantioselective adsorption; penicillamine; cysteine 1. Introduction Chirality has attracted enormous interest in the field of chemistry due to the enantiospecific responses of living organisms to the two enantiomers of chiral compounds that they have ingested. -

Chapter 4: Stereochemistry Introduction to Stereochemistry
Chapter 4: Stereochemistry Introduction To Stereochemistry Consider two of the compounds we produced while finding all the isomers of C7H16: CH3 CH3 2-methylhexane 3-methylhexane Me Me Me C Me H Bu Bu Me Me 2-methylhexane H H mirror Me rotate Bu Me H 2-methylhexame is superimposable with its mirror image Introduction To Stereochemistry Consider two of the compounds we produced while finding all the isomers of C7H16: CH3 CH3 2-methylhexane 3-methylhexane H C Et Et Me Pr Pr 3-methylhexane Me Me H H mirror Et rotate H Me Pr 2-methylhexame is superimposable with its mirror image Introduction To Stereochemistry Consider two of the compounds we produced while finding all the isomers of C7H16: CH3 CH3 2-methylhexane 3-methylhexane .Compounds that are not superimposable with their mirror image are called chiral (in Greek, chiral means "handed") 3-methylhexane is a chiral molecule. .Compounds that are superimposable with their mirror image are called achiral. 2-methylhexane is an achiral molecule. .An atom (usually carbon) with 4 different substituents is called a stereogenic center or stereocenter. Enantiomers Et Et Pr Pr Me CH3 Me H H 3-methylhexane mirror enantiomers Et Et Pr Pr Me Me Me H H Me H H Two compounds that are non-superimposable mirror images (the two "hands") are called enantiomers. Introduction To Stereochemistry Structural (constitutional) Isomers - Compounds of the same molecular formula with different connectivity (structure, constitution) 2-methylpentane 3-methylpentane Conformational Isomers - Compounds of the same structure that differ in rotation around one or more single bonds Me Me H H H Me H H H H Me H Configurational Isomers or Stereoisomers - Compounds of the same structure that differ in one or more aspects of stereochemistry (how groups are oriented in space - enantiomers or diastereomers) We need a a way to describe the stereochemistry! Me H H Me 3-methylhexane 3-methylhexane The CIP System Revisited 1. -

Chirality and Enantiomers
16.11.2010 Bioanalytics Part 5 12.11.2010 Chirality and Enantiomers • Definition: Optical activity • Properties of enantiomers • Methods: – Polarimetry – Circular dichroism • Nomenclature • Separation of enantiomers • Determination of enantiomeric excess (ee) Determination of absolute configuration • Enantiomeric ratio (E‐value) 1 16.11.2010 Definitions Enantiomers: the two mirror images of a molecule Chirality: non‐superimposable mirror‐images •Depends on the symmetry of a molecule •Point‐symmetry: asymmetric C, Si, S, P‐atoms •Helical structures (protein α‐helix) Quarz crystals snail‐shell amino acids Properties of Enantiomers – Chemical identical – Identical UV, IR, NMR‐Spectra – Differences: • Absorption and refraction of circular polarized light is different – Polarimetry, CD‐spectroscopy • Interaction with other chiral molecules/surfaces is different – Separation of enantiomers on chiral columns (GC, HPLC) 2 16.11.2010 Chiral compounds show optical activity A polarimeter is a device which measures the angle of rotation by passing polarized light through an „optical active“ (chiral) substance. Interaction of light and matter If light enters matter, its intensity (amplitude), polarization, velocity, wavelength, etc. may alter. The two basic phenomena of the interaction of light and matter are absorption (or extinction) and a decrease in velocity. 3 16.11.2010 Interaction of light and matter Absorption means that the intensity (amplitude) of light decreases in matter because matter absorbs a part of the light. (Intensity is the square of amplitude.) Interaction of light and matter The decrease in velocity (i.e. the slowdown) of light in matter is caused by the fact that all materials (even materials that do not absorb light at all) have a refraction index, which means that the velocity of light is smaller in them than in vacuum. -

Preferential Crystallization of a Racemic Compound Via Its Conglomerate Co-Crystals
Preferential crystallization of a racemic compound via its conglomerate co-crystals Master Thesis Oscar F. Villamil R August 24th 2016 Faculty of 3ME Department: Process & Energy Section: Intensified Reaction & Separation Systems Graduation Committee Ir. W. Li PDeng Dr. ir. H.J.M Kramer Dr. ir. H.W.Nugteren Dr. ir. A. van der Heijden 1 Abstract Preferential crystallization, as a powerful chiral resolution technique, is intrinsically limited to chiral molecules that crystallize as conglomerates. Many studies have been conducted on using chemical reactions to convert the target molecules, which originally form racemic compounds, into conglomerate-forming derivatives salts or by creating solvate, for the application of preferential crystallization. Up to this date conglomerate co-crystals of racemic compounds have never been applied as the intermediate for chiral resolution. In this study, preferential crystallization of the model compound Ibuprofen (IBU), originally a racemic compound, was carried out via its conglomerate co-crystal with 2,4-bipyridine ethylene (BPE) in heptane. Suitable operation conditions were selected based on pseudo- binary phase diagram of the model compound system constructed under different IBU-BPE ratio. A unique measurement method combining polarimeter and Nuclear Magnetic Resonance (NMR) measurements was developed to identify the enantiopurity and the yield of the final product, which was a mixture of racemic IBU and IBU-BPE co-crystals, a likely result from this complex system. With respect to the results, preferential crystallization of IBU was successfully performed by slowly cooling down a saturated solution of racemic IBU-BPE, initially at T=57.5°C, after seeding it with S-IBU/BPE crystals to T=53°C with a cooling rate of 0.3°C/min. -

Polarimetric Detection of Enantioselective Adsorption by Chiral Au Nanoparticles – Effects of Temperature, Wavelength and Size
Nanomaterials and Nanotechnology ARTICLE Polarimetric Detection of Enantioselective Adsorption by Chiral Au Nanoparticles – Effects of Temperature, Wavelength and Size Invited Article Nisha Shukla2, Nathaniel Ondeck1, Nathan Khosla1, Steven Klara1, Alexander Petti1 and Andrew Gellman1* 1 Department of Chemical Engineering, Carnegie Mellon University, Pittsburgh, PA, USA 2 Institute of Complex Engineered Systems, Carnegie Mellon University, Pittsburgh, PA, USA * Corresponding author(s) E-mail: [email protected] Received 01 November 2014; Accepted 07 January 2015 DOI: 10.5772/60109 © 2015 The Author(s). Licensee InTech. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract 1. Introduction R- and S-propylene oxide (PO) have been shown to interact The importance of chirality arises from the need of the enantiospecifically with the chiral surfaces of Au nanopar‐ pharmaceutical industry, and other producers of bioactive ticles (NPs) modified with D- or L-cysteine (cys). This compounds, for chemical processes that are enantioselec‐ enantiospecific interaction has been detected using optical tive and thereby produce enantiomerically pure chiral polarimetry measurements made on solutions of the D- or products. [1-4] This is one of the most challenging forms of L-cys modified Au (cys/Au) NPs during addition of chemical synthesis and is critical to the performance of racemic PO. The selective adsorption of one enantiomer of bioactive chiral products. Ingestion of the two enantiomers the PO onto the cys/Au NP surfaces results in a net rotation of a chiral compound by a living organism can result in of light during addition of the racemic PO to the solution. -

Optical Rotation and Rotatory Dispersion in Solution and in the Crystalline State
574 consists in a decrease of the number of protons, - that are the particles observed by the counters - partly by ionisation and radiation but also partly being replaced by neutrons which in their turn will be replaced by protons in a lower level. The examination of this absorption process may be decisive for our knowledge of the nature of the most penetrating rays. Chemistry. - Optical Rotation and Rotatory Dispersion in Solution and in the Crystalline State. By F. M. JAEGER, J. TER BERG and P. TERPSTRA. (Communicated at the meeting of June 26, 1937). § 1. If the molecules of a substance possess a dissymmetrical structure and that substance is dissolved in some solvent, the solution will manifest the phenomenon of rotation of the plane of polarization of an incident beam of linearly polarized light. An explanation of this optical rotation was given by FRESNEL in the well-known way by the supposition of the interference of two circularly polarized vibrations of opposite directions. The rotation and the rota tory dispersion are dependent on the temperature, because of the motions of the dissolved molecules being a function of the temperature. In a crystal, the molecules of which oscillate round definite mean positions of equilibrium, there can be two causes of a rotation of the plane of polarization of a polarized light-beam traversing the crystal: 1°. Either the molecules composing the crystal are themselves symmetrical and, therefore, optically inactive, - but their arrangement in the crystalstructure is a dissymmetrical one (quartz; sodiumchlorate). The optical activity of the crystal then is an immediate consequence of that dissymmetrical arrangement and it vanishes as soon as the latter is destroyed, - for instance by dissolving or melting the crystal. -

A Review on Chiral Chromatography and Its Application to the Pharmaceutical Industry
Chemsearch Journal 2(1): 8 - 11 Publication of Chemical Society of Nigeria, Kano Chapter CHIRAL CHROMATOGRAPHY AND ITS APPLICATION TO THE PHARMACEUTICAL INDUSTRY: A REVIEW Mudi, S. Y. and *Muhammad, A. Department of Pure and Industrial Chemistry, Bayero University, PMB 3011, Kano. *Correspondence author: [email protected] ABSTRACT Chiral chromatographic enantioseparation has been in practice by researchers. There has been a considerable interest in the synthesis and separation of enantiomers of organic compounds especially because of their importance in the biochemical and pharmaceutical industries. Often, these compounds are purified rather than being produced by chiral-specific synthesis. We herein present a general discussion that focuses on the chromatographic enantioseparation, which we hope will be useful to chromatographic and pharmaceutical industries. Keywords: Chiral chromatography, enantioseparation, pharmaceutical industry. INTRODUCTION giving differing affinities between the analytes Chromatography is the collective term for a set of (Schreier et al., 1995). laboratory techniques for the separation of mixtures. It The main goal of this review is to provide a brief involves passing a mixture dissolved in a "mobile overview of chiral separations to researchers who phase" through a “stationary phase”, which separates are versed in the area of analytical separations but the analyte from other compounds in the mixture unfamiliar with chiral separations. This review based on differential partitioning between the mobile highlights significant issues of the chiral and stationary phases. Subtle differences in a separations and provides salient examples from compound's partition coefficient result in differential specific classes of chiral selectors where retention on the stationary phase and thus effecting appropriate. the separation (Laurence and Christopher, 1989; Pascal et al., 2000). -

1 the Historical Development of Asymmetric Hydrogenation
1 1 The Historical Development of Asymmetric Hydrogenation John M. Brown Chemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford OX1 3TA, UK 1.1 Introduction How did chemists gain the current levels of knowledge and expertise for control- ling molecular chirality through hydrogenation or otherwise? The desirability of asymmetric synthesis was recognized in the 1880s by Emil Fischer and others, but practical solutions only arose more than 80 years later. The key reasons are explored here. This brief review has five main Sections 1.2–1.6, covering first the development of ideas underpinning our understanding of asymmetry, then the initial applications to asymmetric synthesis, and also the development of asymmetric heterogeneous hydrogenation of alkenes. The final sections on asymmetric homogeneous hydro- genation of alkenes are limited to work published in or before the early 1980s, in advance of extensive developments, and thus excluding the important inputs of irid- ium catalysts and more recently early transition metals. 1.2 Early Work on the Recognition of Molecular Asymmetry Chemistry was an emerging science by the beginning of the nineteenth century with many opportunities for fundamental discovery. At that time scientists crossed dis- ciplines easily; optics and mineralogy played important roles because of the ready accessibility and verifiable purity of solid substances. Malus had invented the first polarimeter in 1808, enabling measurement of both the sense and magnitude of rota- tion of plane-polarized light [4]. Following this, work by Arago and others on the interaction of polarized light with minerals intensified in the following decade [5]. Haüy had earlier concluded that each type of crystal has a fundamental primitive, nucleus or “integrant molecule” of a particular shape, that could not be broken fur- ther without destroying both the physical and chemical nature of the crystal. -

Optical Rotation
Optical Rotation Classical Interpretation Optically active substances can rotate the plane of polarization of light that passes through them. From the classical viewpoint this requires (1) that the oscillating magnetic field of the incident light induce an electric dipole moment in the molecule parallel to the incident magnet field and (2) that the electric field of the incident light must induce a corresponding magnetic moment parallel to the incident electric field. Thus, where m and y are the induced electric and magnetic dipole moments and H and E are the magnetic and electric fields of the incident light. There is a simple qualitative interpretation of the significance of these requirements, as shown in Figures 15-24 and 15-25 from Kauzmann's Ouantum Chemistrv, below. Consider a molecule in which the electrons are constrained to move along a helical path. Suppose plane polarized light is incident upon the molecule such that the oscillating magnetic field is parallel to the helix axis. Then according to Faraday's laws the changing magnetic field induces an EMF in the helical conducting path, and so causes a resultant oscillating electric dipole parallel to the helix axis, i.e., parallel to the magnetic field of the incident light. This oscillating electric dipole produces an oscillating electric field which is perpendicular to the electric field of the incident light. Since the incident and induced fields are in phase, they can be combined by vector addition, leading to a resultant electric field which is rotated relative to the incident field. The same argument holds for the magnetic field induced by the alternating electric field of the light. -

The Optical Rotation of Liquids Its Variation with Wave Length Temperature, Solvent and Concentration
U. S. DEPARTMENT OF COMMERCE BUREAU OF STANDARDS THE OPTICAL ROTATION OF LIQUIDS ITS VARIATION WITH WAVE LENGTH TEMPERATURE, SOLVENT AND CONCENTRATION By T. MARTIN LOWRY MISCELLANEOUS PUBLICATION No. 118 U. S. DEPARTMENT OF COMMERCE R. P. LAMONT, Secretary BUREAU OF STANDARDS GEORGE K. BURGESS, Director Miscellaneous Publication No. 118 THE OPTICAL ROTATION OF LIQUIDS ITS VARIATION WITH WAVE LENGTH TEMPERATURE, SOLVENT AND CONCENTRATION BY T. MARTIN LOWRY MARCH 25, 1932 UNITED STATES GOVERNMENT PRINTING OFFICE WASHINGTON : 1932 For sale by the superintendent of Documents, Washington, d. c. PREFACE The compilation of the available data on this topic, as presented in the following pages, was made in conjunction with work undertaken for the International Critical Tables and covers, with the exceptions noted below, the literature preceding January 1, 1923. No attempt has been made to include fragmentary data. Data for the sodium D-line at ordinary temperatures and in the common solvents will be found in International Critical Tables. 1 The common sugars have also been omitted from the present compilation, since they have been covered in the saccharimetry section of International Critical Tables. 2 i Int. Crit. Tables, 7, pp. 355-489. McGraw-Hill Book Co., New York; 1931. » Int. Crit. Tables, 2, pp. 334-355. McGraw-Hill Book Co., New York; 1927. CONTENTS Page Preface n L Arrangement 1 II. Symbols and abbreviations 2 III. Class I. Organic substances in which the asymmetric carbon atom does not form part of a ring 2 IB. The molecule contains one carbon atom attached to four different atoms or groups 2 IBi. -

Selective Chiral Symmetry Breaking: When “Left” and “Right” Cannot Coexist and “Left” Is the Best Option
1 Selective chiral symmetry breaking: when “Left” and “Right” cannot coexist and “Left” is the best option. Cristobal Viedma Department of Crystallography and Mineralogy, Faculty of Geology, Complutense University, Madrid, Spain. E-mail: [email protected] Chiral symmetry breaking occurs when a physical or chemical process, with no preference for the production of one or other enantiomer, spontaneously generates a large excess of one of the two enantiomers: (L), left-handed or (D), right handed. Inorganic processes involving chiral products commonly yield a racemic mixture of both. However life on Earth uses only one type of amino acids (L) and one type of natural sugars (D). The origin of this selective chirality has remained a fundamental enigma in the origin of life since the time of Pasteur, some 140 years ago. Sodium bromate (NaBrO 3 ) and sodium chlorate (NaClO 3 ) when crystallize from an unstirred solution generates statistically equal numbers of left-handed (L) and right handed (D) chiral crystals. But when these two populations of crystals undergo a dissolution-crystallization phenomenon, they cannot coexist: one of them disappears in an irreversible competition process that nurtures the other one. From the viewpoint of energy, these two enantiomers can exist with an equal probability, thus the result of this competition in different systems would be populations of crystals either (L) or (D). But contradicting this theoretical prediction the handedness of the chiral crystals that win the competition and remain in solution in the different systems is almost always the same (99.2 percent for NaBrO 3 ). We suggest that these results are the consequence of Parity-violating energy difference (PVED) between enantiomers and reinforce the idea of a key role of PVDE theory in the origin of biomolecular chirality on Earth. -
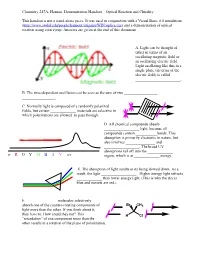
Optical Rotation Demonstration Handout
Chemistry 247A, Hanson. Demonstration Handout—Optical Rotation and Chirality This handout is not a stand-alone piece. It was used in conjunction with a Visual Basic 6.0 simulation (http://www.stolaf.edu/people/hansonr/origami/WIN/optics.exe) and a demonstration of optical rotation using corn syrup. Answers are given at the end of this document. A. Light can be thought of either in terms of an oscillating magnetic field or an oscillating electric field. Light oscillating like this in a single plane (in terms of the electric field) is called _______________________. B. The time-dependent oscillation can be seen as the sum of two _________________ _________________. C. Normally light is composed of a randomly polarized fields, but certain _____________ materials are selective in which polarizations are allowed to pass through. D. All chemical compounds absorb __________________ light, because all compounds contain ___________bonds. This absorption is primarily electronic in nature, but also involves _______________ and __________________. The broad UV absorptions tail off into the _____________ ir R O Y G B I V uv region, which is at _____________ energy. E. The absorption of light results in its being slowed down. As a result, the light __________________. Higher energy light refracts _____________ than lower energy light. (This is why the sky is blue and sunsets are red.) F. _______________ molecules selectively absorb one of the counter-rotating components of Br CH3 light more than the other. If you think about it, they have to. How could they not? This H Cl ―retardation‖ of one component more than the other results in a rotation of the plane of polarization.