Rare Coagulopathies Chapter 5 Author
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Factor XIII and Fibrin Clot Properties in Acute Venous Thromboembolism
International Journal of Molecular Sciences Review Factor XIII and Fibrin Clot Properties in Acute Venous Thromboembolism Michał Z ˛abczyk 1,2 , Joanna Natorska 1,2 and Anetta Undas 1,2,* 1 John Paul II Hospital, 31-202 Kraków, Poland; [email protected] (M.Z.); [email protected] (J.N.) 2 Institute of Cardiology, Jagiellonian University Medical College, 31-202 Kraków, Poland * Correspondence: [email protected]; Tel.: +48-12-614-30-04; Fax: +48-12-614-21-20 Abstract: Coagulation factor XIII (FXIII) is converted by thrombin into its active form, FXIIIa, which crosslinks fibrin fibers, rendering clots more stable and resistant to degradation. FXIII affects fibrin clot structure and function leading to a more prothrombotic phenotype with denser networks, characterizing patients at risk of venous thromboembolism (VTE). Mechanisms regulating FXIII activation and its impact on fibrin structure in patients with acute VTE encompassing pulmonary embolism (PE) or deep vein thrombosis (DVT) are poorly elucidated. Reduced circulating FXIII levels in acute PE were reported over 20 years ago. Similar observations indicating decreased FXIII plasma activity and antigen levels have been made in acute PE and DVT with their subsequent increase after several weeks since the index event. Plasma fibrin clot proteome analysis confirms that clot-bound FXIII amounts associated with plasma FXIII activity are decreased in acute VTE. Reduced FXIII activity has been associated with impaired clot permeability and hypofibrinolysis in acute PE. The current review presents available studies on the role of FXIII in the modulation of fibrin clot properties during acute PE or DVT and following these events. -

MYH9-Related Platelet Disorders
Reprinted with permission from Thieme Medical Publishers (Semin Thromb Hemost 2009;35:189-203) Homepage at www.thieme.com MYH9-Related Platelet Disorders Karina Althaus, M.D.,1 and Andreas Greinacher, M.D.1 ABSTRACT Myosin heavy chain 9 (MYH9)-related platelet disorders belong to the group of inherited thrombocytopenias. The MYH9 gene encodes the nonmuscle myosin heavy chain IIA (NMMHC-IIA), a cytoskeletal contractile protein. Several mutations in the MYH9 gene lead to premature release of platelets from the bone marrow, macro- thrombocytopenia, and cytoplasmic inclusion bodies within leukocytes. Four overlapping syndromes, known as May-Hegglin anomaly, Epstein syndrome, Fechtner syndrome, and Sebastian platelet syndrome, describe different clinical manifestations of MYH9 gene mutations. Macrothrombocytopenia is present in all affected individuals, whereas only some develop additional clinical manifestations such as renal failure, hearing loss, and presenile cataracts. The bleeding tendency is usually moderate, with menorrhagia and easy bruising being most frequent. The biggest risk for the individual is inappropriate treatment due to misdiagnosis of chronic autoimmune thrombocytopenia. To date, 31 mutations of the MYH9 gene leading to macrothrombocytopenia have been identified, of which the upstream mutations up to amino acid 1400 are more likely associated with syndromic manifestations than the downstream mutations. This review provides a short history of MYH9-related disorders, summarizes the clinical and laboratory character- istics, describes a diagnostic algorithm, presents recent results of animal models, and discusses aspects of therapeutic management. KEYWORDS: MYH9 gene, nonmuscle myosin IIA, May-Hegglin anomaly, Epstein syndrome, Fechtner syndrome, Sebastian platelet syndrome, macrothrombocytopenia The correct diagnosis of hereditary chronic as isolated platelet count reductions or as part of thrombocytopenias is important for planning appropri- more complex clinical syndromes. -

Factor XIII Deficiency
Factor XIII deficiency Information for families Great Ormond Street Hospital for Children NHS Foundation Trust 2 Factor XIII deficiency is a type of clotting disorder. A specific protein is missing from the blood so that injured blood vessels cannot heal in the usual way. This information sheet from Great Ormond Street Hospital (GOSH) explains the causes, symptoms and treatment of Factor XIII deficiency and where to get help. What is a clotting disorder? A clotting (or coagulation) disorder is a on in order. When all of the factors are turned medical condition where a specific protein on, the blood forms a clot which stops the is missing from the blood. injury site bleeding any further. Blood is made up of different types of There are a number of coagulation factors cells (red blood cells, white blood cells and circulating in the blood, lying in wait to be platelets) all suspended in a straw-coloured turned on when an injury occurs. If any one liquid called plasma. Platelets are the cells of the factors is missing from the body, the responsible for making blood clot. When complicated chemical reaction described a blood vessel is injured, platelets clump above will not happen as it should. This can together to block the injury site. They also lead to blood loss, which can be severe and start off a complicated chemical reaction to life-threatening. Each coagulation factor form a mesh made of a substance called fibrin. is given a number from I to XIII – they are This complicated chemical reaction always always written as Roman numerals – and follows a strict pattern – with each clotting the effects of the missing factor will vary. -

The Rare Coagulation Disorders
Treatment OF HEMOPHILIA April 2006 · No. 39 THE RARE COAGULATION DISORDERS Paula HB Bolton-Maggs Department of Haematology Manchester Royal Infirmary Manchester, United Kingdom Published by the World Federation of Hemophilia (WFH) © World Federation of Hemophilia, 2006 The WFH encourages redistribution of its publications for educational purposes by not-for-profit hemophilia organizations. In order to obtain permission to reprint, redistribute, or translate this publication, please contact the Communications Department at the address below. This publication is accessible from the World Federation of Hemophilia’s web site at www.wfh.org. Additional copies are also available from the WFH at: World Federation of Hemophilia 1425 René Lévesque Boulevard West, Suite 1010 Montréal, Québec H3G 1T7 CANADA Tel. : (514) 875-7944 Fax : (514) 875-8916 E-mail: [email protected] Internet: www.wfh.org The Treatment of Hemophilia series is intended to provide general information on the treatment and management of hemophilia. The World Federation of Hemophilia does not engage in the practice of medicine and under no circumstances recommends particular treatment for specific individuals. Dose schedules and other treatment regimes are continually revised and new side effects recognized. WFH makes no representation, express or implied, that drug doses or other treatment recommendations in this publication are correct. For these reasons it is strongly recommended that individuals seek the advice of a medical adviser and/or to consult printed instructions provided by the pharmaceutical company before administering any of the drugs referred to in this monograph. Statements and opinions expressed here do not necessarily represent the opinions, policies, or recommendations of the World Federation of Hemophilia, its Executive Committee, or its staff. -

Familial Multiple Coagulation Factor Deficiencies
Journal of Clinical Medicine Article Familial Multiple Coagulation Factor Deficiencies (FMCFDs) in a Large Cohort of Patients—A Single-Center Experience in Genetic Diagnosis Barbara Preisler 1,†, Behnaz Pezeshkpoor 1,† , Atanas Banchev 2 , Ronald Fischer 3, Barbara Zieger 4, Ute Scholz 5, Heiko Rühl 1, Bettina Kemkes-Matthes 6, Ursula Schmitt 7, Antje Redlich 8 , Sule Unal 9 , Hans-Jürgen Laws 10, Martin Olivieri 11 , Johannes Oldenburg 1 and Anna Pavlova 1,* 1 Institute of Experimental Hematology and Transfusion Medicine, University Clinic Bonn, 53127 Bonn, Germany; [email protected] (B.P.); [email protected] (B.P.); [email protected] (H.R.); [email protected] (J.O.) 2 Department of Paediatric Haematology and Oncology, University Hospital “Tzaritza Giovanna—ISUL”, 1527 Sofia, Bulgaria; [email protected] 3 Hemophilia Care Center, SRH Kurpfalzkrankenhaus Heidelberg, 69123 Heidelberg, Germany; ronald.fi[email protected] 4 Department of Pediatrics and Adolescent Medicine, University Medical Center–University of Freiburg, 79106 Freiburg, Germany; [email protected] 5 Center of Hemostasis, MVZ Labor Leipzig, 04289 Leipzig, Germany; [email protected] 6 Hemostasis Center, Justus Liebig University Giessen, 35392 Giessen, Germany; [email protected] 7 Center of Hemostasis Berlin, 10789 Berlin-Schöneberg, Germany; [email protected] 8 Pediatric Oncology Department, Otto von Guericke University Children’s Hospital Magdeburg, 39120 Magdeburg, Germany; [email protected] 9 Division of Pediatric Hematology Ankara, Hacettepe University, 06100 Ankara, Turkey; Citation: Preisler, B.; Pezeshkpoor, [email protected] B.; Banchev, A.; Fischer, R.; Zieger, B.; 10 Department of Pediatric Oncology, Hematology and Clinical Immunology, University of Duesseldorf, Scholz, U.; Rühl, H.; Kemkes-Matthes, 40225 Duesseldorf, Germany; [email protected] B.; Schmitt, U.; Redlich, A.; et al. -

Intracerebral Bleeding in Young Infants Due to Rare Etiologies–A
eona f N tal l o B a io n l r o u g y o J Tewari et al., J Neonatal Biol 2016, 5:2 Journal of Neonatal Biology DOI: 10.4172/2167-0897.1000221 ISSN: 2167-0897 Case Report Open access Intracerebral Bleeding in Young Infants due to Rare Etiologies–A Report of two Cases Vishal Vishnu Tewari1*, Kunal Tewari2 and Ritu Mehta3 1Department of Pediatrics, Army Hospital (Referral and Research), New Delhi, India 2Department of Anaesthesia, Classified Specialist Anesthesia, Base Hospital, New Delhi, India 3Department of Pathology, All India Institute of Medical Sciences, New Delhi, India *Corresponding author: Vishal Vishnu Tewari, Department of Pediatrics, Army Hospital, New Delhi, India,Tel: +91-8826118889, E-mail: [email protected] Rec date: April 29, 2016; Acc date: May 16, 2016; Pub date: May 23, 2016 Copyright: © 2016 Tewari VV, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract Neonates and young infants are physiologically encumbered by an inadequate hemostatic mechanism. They may also have inherited or acquired bleeding disorders which may have a catastrophic presentation with intracerebral hemorrhage. The need to reach an accurate diagnosis is paramount in order to provide accurate therapy and genetic counseling. We report two infants who presented with unprovoked life threatening massive intracerebral hemorrhage. The first infant was diagnosed and managed for congenital factor V deficiency while the second infant had Glanzmann thrombasthenia. Keywords: Intracerebral bleeding; Congenital factor V deficiency; disorder in the family. -

Familial Haemophilia and Factor Vii Deficiency by M
J Clin Pathol: first published as 10.1136/jcp.11.5.412 on 1 September 1958. Downloaded from J. clin. Path. (1958), 11, 412. FAMILIAL HAEMOPHILIA AND FACTOR VII DEFICIENCY BY M. CONSTANDOULAKIS Front the Group Laboratory, St. Mary Abbots Hospital, London* (RECEIVED FOR PUBLICATION MAY 12, 1958) This investigation is reported because of the months later he was readmitted with a few bruises combination of familial haemophilia and factor on the legs and bleeding gums. Bleeding subsided with VII deficiency and the unusual occurrence of a local treatment and vitamin K1. female haemophiliac. Brenda, his sister (aged 10), appears normal with- Cases of combined deficiencies of different out any haemorrhagic manifestations up till now. factors are extremely rare and up till now Eileen Br., his mother (aged 46), revealed that she clotting had had excessive bleeding during both her deliveries, there have been reported combinations of haemo- during and after a hysterectomy, being transfused in philia and factor V deficiency (Koller, 1954), order to control haemorrhage, and after tooth extrac- haemophilia and Christmas disease (Soulier and tions at different times. Larrieu, 1953), Christmas disease and factor VII Frank G. (aged 43), his maternal uncle, has bled deficiency (Bell and Alton, 1955; Stein and since a small child after trauma and tooth extrac- Abrahams, 1956; de Vries, Kettenborg, and van tions, oozing from sockets usually persisting for about der Pol, 1955), but not of haemophilia and factor three days. In 1948 he had a blow in the abdomencopyright. VII deficiency. which was followed by severe haematemesis and Female haemophiliacs in the homozygous state melaena and he was transfused in order to control have been reported by Merskey (1951), Israels, the haemorrhage. -
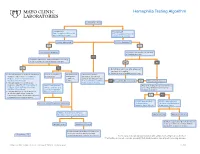
Hemophilia Testing Algorithm
Hemophilia Testing Algorithm Symptomatic male Hemophilia A Hemophilia B F8A / Coagulation Factor VIII F_9 / Coagulation Factor IX Activity Assay, Plasma Activity Assay, Plasma Activity decreased? Activity decreased? YES YES Hemophilia A diagnosis ■ Vitamin K antagonist (ie, warfarin) ■ Child/adolescent? NO F8 genetic testing has been performed on a family member and the specific mutation is known? YES NO YES NO ■ Retest when >4 weeks after antagonist treatment is complete ■ Adjust decreased activity levels for age ■ If known mutation is an Intron 1 Inversion Severe hemophilia: Moderate/mild If the activity assays mutation, order F81B / Hemophilia A activity <1% hemophilia: are normal, consider an F8 Gene, Intron 1 Inversion Known activity 1% alternate bleeding disorder: Mutation, Whole Blood* to 55% ALBLD / Bleeding Diathesis NO Is activity still decreased? YES Hemophilia B diagnosis ■ If known mutation is an Intron 22 Profile, Limited, Plasma inversion, order F822B / Hemophilia A F8INV / Hemophilia A F9 genetic testing has been performed F8 Gene, Intron 22 Inversion Known F8 Gene, Intron 1 and on a family member and the specific Mutation, Whole Blood* 22 Inversion Mutation mutation is known? ■ If known mutation is a point mutation Analysis, Whole Blood or deletion/duplication, contact a Laboratory Genetic Counselor to discuss YES NO targeted familial mutation testing FIXKM / Hemophilia B, NGSF9 / Hemophilia B, Inversion found Inversion not found F9 Gene Known F9 Gene, Next-Generation Mutation, Whole Blood* Sequencing, Varies F8NGS / -

Coagulation Factors Directly Cleave SARS-Cov-2 Spike and Enhance Viral Entry
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.31.437960; this version posted April 1, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Coagulation factors directly cleave SARS-CoV-2 spike and enhance viral entry. Edward R. Kastenhuber1, Javier A. Jaimes2, Jared L. Johnson1, Marisa Mercadante1, Frauke Muecksch3, Yiska Weisblum3, Yaron Bram4, Robert E. Schwartz4,5, Gary R. Whittaker2 and Lewis C. Cantley1,* Affiliations 1. Meyer Cancer Center, Department of Medicine, Weill Cornell Medical College, New York, NY, USA. 2. Department of Microbiology and Immunology, Cornell University, Ithaca, New York, USA. 3. Laboratory of Retrovirology, The Rockefeller University, New York, NY, USA. 4. Division of Gastroenterology and Hepatology, Department of Medicine, Weill Cornell Medicine, New York, NY, USA. 5. Department of Physiology, Biophysics and Systems Biology, Weill Cornell Medicine, New York, NY, USA. *Correspondence: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2021.03.31.437960; this version posted April 1, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Summary Coagulopathy is recognized as a significant aspect of morbidity in COVID-19 patients. The clotting cascade is propagated by a series of proteases, including factor Xa and thrombin. Other host proteases, including TMPRSS2, are recognized to be important for cleavage activation of SARS-CoV-2 spike to promote viral entry. Using biochemical and cell-based assays, we demonstrate that factor Xa and thrombin can also directly cleave SARS-CoV-2 spike, enhancing viral entry. -

Diagnosis of Inherited Platelet Disorders on a Blood Smear
Journal of Clinical Medicine Article Diagnosis of Inherited Platelet Disorders on a Blood Smear Carlo Zaninetti 1,2,3 and Andreas Greinacher 1,* 1 Institut für Immunologie und Transfusionsmedizin, Universitätsmedizin Greifswald, 17489 Greifswald, Germany; [email protected] 2 University of Pavia, and IRCCS Policlinico San Matteo Foundation, 27100 Pavia, Italy 3 PhD Program of Experimental Medicine, University of Pavia, 27100 Pavia, Italy * Correspondence: [email protected]; Tel.: +49-3834-865482; Fax: +49-3834-865489 Received: 19 January 2020; Accepted: 12 February 2020; Published: 17 February 2020 Abstract: Inherited platelet disorders (IPDs) are rare diseases featured by low platelet count and defective platelet function. Patients have variable bleeding diathesis and sometimes additional features that can be congenital or acquired. Identification of an IPD is desirable to avoid misdiagnosis of immune thrombocytopenia and the use of improper treatments. Diagnostic tools include platelet function studies and genetic testing. The latter can be challenging as the correlation of its outcomes with phenotype is not easy. The immune-morphological evaluation of blood smears (by light- and immunofluorescence microscopy) represents a reliable method to phenotype subjects with suspected IPD. It is relatively cheap, not excessively time-consuming and applicable to shipped samples. In some forms, it can provide a diagnosis by itself, as for MYH9-RD, or in addition to other first-line tests as aggregometry or flow cytometry. In regard to genetic testing, it can guide specific sequencing. Since only minimal amounts of blood are needed for the preparation of blood smears, it can be used to characterize thrombocytopenia in pediatric patients and even newborns further. -

ISTH Couverture 6.6.2012 10:21 Page 1 ISTH Couverture 6.6.2012 10:21 Page 2 ISTH Couverture 6.6.2012 10:21 Page 3 ISTH Couverture 6.6.2012 10:21 Page 4
ISTH Couverture 6.6.2012 10:21 Page 1 ISTH Couverture 6.6.2012 10:21 Page 2 ISTH Couverture 6.6.2012 10:21 Page 3 ISTH Couverture 6.6.2012 10:21 Page 4 ISTH 2012 11.6.2012 14:46 Page 1 Table of Contents 3 Welcome Message from the Meeting President 3 Welcome Message from ISTH Council Chairman 4 Welcome Message from SSC Chairman 5 Committees 7 ISTH Future Meetings Calendar 8 Meeting Sponsors 9 Awards and Grants 2012 12 General Information 20 Programme at a Glance 21 Day by Day Scientific Schedule & Programme 22 Detailed Programme Tuesday, 26 June 2012 25 Detailed Programme Wednesday, 27 June 2012 33 Detailed Programme Thursday, 28 June 2012 44 Detailed Programme Friday, 29 June 2012 56 Detailed Programme Saturday, 30 June 2012 68 Hot Topics Schedule 71 ePoster Sessions 97 Sponsor & Exhibitor Profiles 110 Exhibition Floor Plan 111 Congress Centre Floor Plan www.isth.org ISTH 2012 11.6.2012 14:46 Page 2 ISTH 2012 11.6.2012 14:46 Page 3 WelcomeCommittees Messages Message from the ISTH SSC 2012 Message from the ISTH Meeting President Chairman of Council Messages Dear Colleagues and Friends, Dear Colleagues and Friends, We warmly welcome you to the elcome It is my distinct privilege to welcome W Scientific and Standardization Com- you to Liverpool for our 2012 SSC mittee (SSC) meeting of the Inter- meeting. national Society on Thrombosis and Dr. Cheng-Hock Toh and his col- Haemostasis (ISTH) at Liverpool’s leagues have set up a great Pro- UNESCO World Heritage Centre waterfront! gramme aiming at making our off-congress year As setting standards is fundamental to all quality meeting especially attractive for our participants. -

Whole-Exome Sequencing of a Patient with Severe and Complex Hemostatic Abnormalities Reveals a Possible Contributing Frameshift Mutation in C3AR1
Downloaded from molecularcasestudies.cshlp.org on October 2, 2021 - Published by Cold Spring Harbor Laboratory Press Whole-exome sequencing of a patient with severe and complex hemostatic abnormalities reveals a possible contributing frameshift mutation in C3AR1 Eva Leinøe1, Ove Juul Nielsen1, Lars Jønson2 and Maria Rossing2∗ Department of Hematology1 and Center for Genomic Medicine2, Rigshospitalet, University of Copenhagen, Blegdamsvej 9, DK-2100 Copenhagen, Denmark Running head: WES reveals a C3AR1 mutation in a complex hemostatic patient ∗Corresponding author: Maria Rossing Center for Genomic Medicine Rigshospitalet University of Copenhagen Blegdamsvej 9 DK-2100 Copenhagen Denmark E-mail: [email protected] Phone: +45 3545 3016 Fax: +45 3545 4435 1 Downloaded from molecularcasestudies.cshlp.org on October 2, 2021 - Published by Cold Spring Harbor Laboratory Press Abstract The increasing availability of genome-wide analysis has made it possible to rapidly sequence the exome of patients with undiagnosed or unresolved medical conditions. Here, we present the case of a 64-year-old male patient with schistocytes in the peripheral blood smear and a complex and life-threatening coagulation disorder causing recurrent venous thromboembolic events, severe thrombocytopenia, and subdural hematomas. Whole-exome sequencing revealed a frameshift mutation (C3AR1 c.355-356dup, p.Asp119Alafs*19) resulting in a premature stop in C3AR1 (Complement Component 3a Receptor 1). Based on this finding, atypical hemolytic uremic syndrome was suspected due to a genetic predisposition, and a targeted treatment regime with Eculizumab was initiated. Life-threatening hemostatic abnormalities would most likely have persisted had it not been for the implementation of whole-exome sequencing in this particular clinical setting.