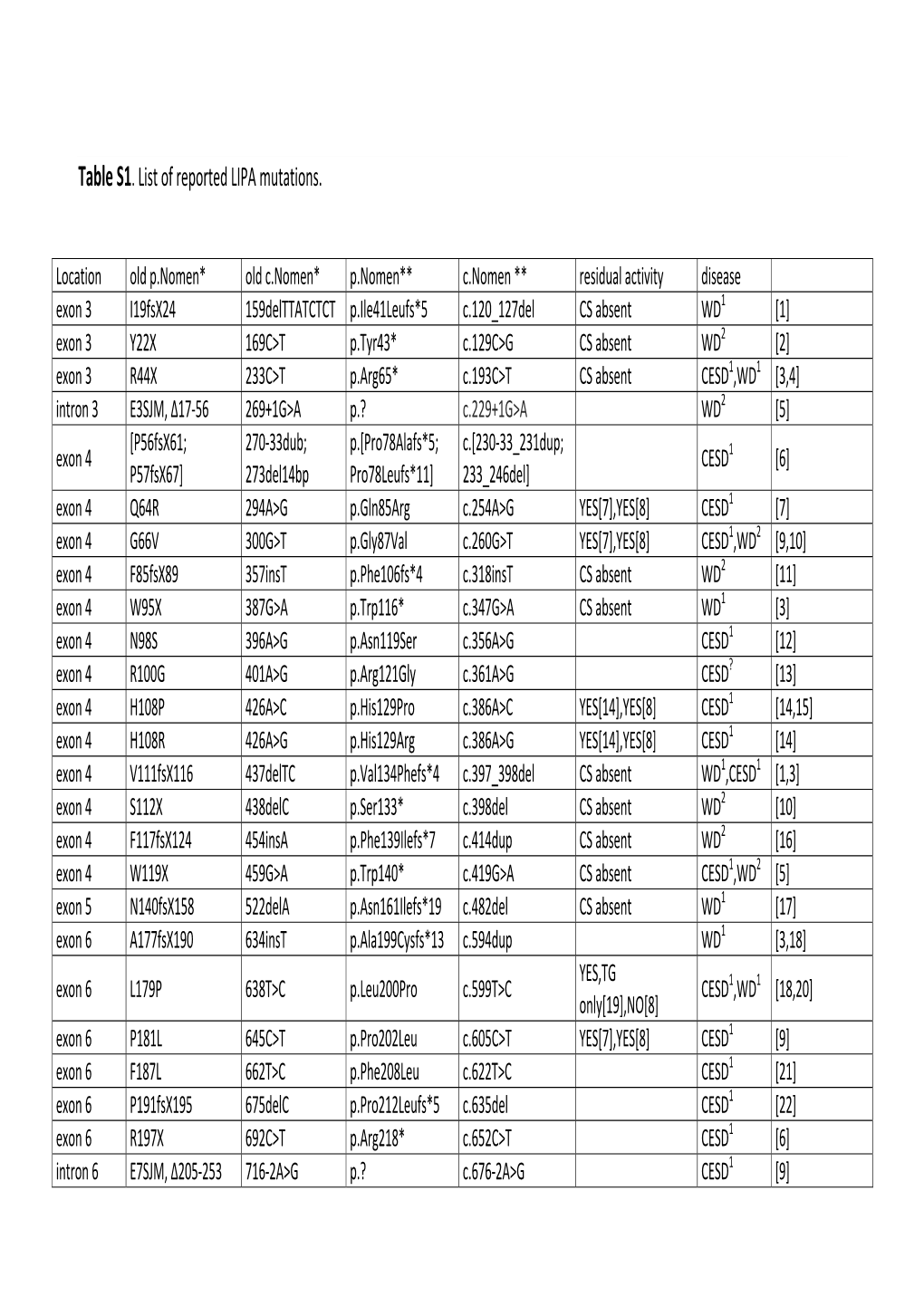
Table S1. List of Reported LIPA Mutations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Autophagy As a Promoter of Longevity – Insights from Model Organisms
1 2 3 4 Autophagy as a promoter of longevity – insights from model organisms 5 Malene Hansen1, David C. Rubinsztein2,3, and David W. Walker4,5 6 7 8 1Sanford Burnham Prebys Medical Discovery Institute, Program of Development, Aging and 9 Regeneration, 10901 North Torrey Pines Road, La Jolla, CA 92037, USA. 10 2Cambridge Institute for Medical Research, Department of Medical Genetics; 11 3UK Dementia Research Institute, University of Cambridge, Hills Road, Cambridge CB2 0XY, 12 UK. 13 4 Department of Integrative Biology and Physiology, University of California, Los Angeles, CA 14 90095, USA; 15 5Molecular Biology Institute, University of California, Los Angeles, CA 90095, USA. 16 17 Correspondence: 18 MH ([email protected]), DWW ([email protected]), DCR ([email protected]) 19 20 Keywords: 21 Selective autophagy, tissue specificity, pathophysiology, ageing, S. cerevisiae, C. elegans, 22 Drosophila 23 24 1 25 Glossary 26 Aggrephagy: The selective removal of cytosolic aggregates by autophagy. 27 28 Autophagosome: A cytosolic double membrane-bound vesicle, capable of sequestering 29 cytoplasmic inclusions and organelles destined for degradation in the autolysosome. 30 31 Autolysosome: A cytosolic vesicle resulting from fusion between an autophagosome and 32 acidic lysosomes in which degradation of the inner membrane and sequestered material in the 33 autophagosome takes place. 34 35 Glomerulus: A key structure of a nephron, the functional unit of the kidney. 36 37 Hormesis/Hormetic heat shock: Beneficial effects of a treatment that at a higher intensity is 38 harmful. In one form of hormesis, non-lethal exposure to elevated temperature induces a 39 response that results in increased stress resistance and longevity. -

Scientific Report 2012 Ongoing Research 2013
SCIENTIFIC REPORT 2012 ONGOING RESEARCH 2013 IRCCS “Istituto Giannina Gaslini” Via Gerolamo Gaslini, 5 16147 Genova – Italy Tel. +39 010 5636 806/807 Fax +39 010 3776590 e-mail: [email protected] www.gaslini.org Some pictures of the Istituto Giannina Gaslini Nobel laureates at Gaslini: Renato Dulbecco and Rolf Zinkernagel Pope Benedict XVI visiting Gaslini Monsignor Angelo Bagnasco visiting Gaslini Some moments of the visit of the International Scientific Committee (Professors Alain Fischer, Max Cooper, Sergio Romagnani and Anthony Fauci) Annual Meeting of the SIOP Brain Tumor Sub-Committee The Germana International Centre for Studies and training (CISEF): a center of excellence which carries out educational activities in the fields of scientific research, prediatrics, organization and quality of health care services. TRIPR (Translational Research in Pediatric Rheumatology) Congress 2009 The 2 nd Training Course on Blood and Marrow Trasplantation: a course of paediatricians and pediatric nurses on HSCT in children and adolescents “I am not a man of science, but I am perfectly aware that only by starting from scientific research, conducted under proper direction, can physicians conscientiously accomplish their difficult task” (Gerolamo Gaslini) Foreword A recent publication by Via Academy - “Small is beautiful” – analyzed the quality of research carried out in different Italian universities and institutes. Small and medium-sized organizations with fewer than 100 principal investigators (PI) or university professors were extracted and in this ranking, based on the ratio of PI/TIS (Top Italian Scientists, Via Academy), Gaslini is ranked first, closely above the Humanitas Institute of Milan. This is a remarkable result and underscores once more the caliber of Gaslini’s researchers, among whom 23 are TIS, equally distributed between basic and clinical investigators. -

The Activity of Lysosomal Enzymes in Blood Serum, Liver, Kidney and Skeletal Muscle of Rabbits Divergently Selected for Locomotor Activity in Open-Field Test
Animal Science Papers and Reports vol. 27 (2009) no. 3, 249-257 Institute of Genetics and Animal Breeding, Jastrzębiec, Poland REPORT The activity of lysosomal enzymes in blood serum, liver, kidney and skeletal muscle of rabbits divergently selected for locomotor activity in open-field test Artur Jóźwik*, Anna Śliwa-Jóźwik, Tadeusz Jezierski, Wojciech Daniewski, Aleksandra Górecka, Adam Kołątaj Polish Academy of Sciences Institute of Genetics and Animal Breeding, Jastrzębiec, 05-552 Wólka Kosowska, Poland (Received August 25, 2008; accepted March 10, 2009) Twenty males were used from generation 8 of rabbits divergently selected for high (line H, n=10) vs. low (line L, n=10) locomotor activity in the open-field (OF-test). At the age of 98 days, blood samples were withdrawn from the ear vein and the rabbits were killed. Immediately, liver, kidney and the left femoral muscle samples were excised. In lysosomal fractions of blood serum and three tissues mentioned the activity of lysosomal enzymes AlaAP, LeuAP, ArgAP, Cat D and L, AcP, EL, LAL, BGRD, BGAL, BGLU, aGlu, MAN and HEX was determined. Significant interline differences were identified in activity of blood and tissue enzymes in question. The results suggest that the divergent selection for high vs. low locomotor activity in the open-field (OF-test) alters the lysosomal degradation processes in the organism. KEY WORDS: lysosomal enzymes / open-field test / rabbits / selection *Corresponding author: [email protected] 249 A. Jóźwik et al. The metabolic processes in live cells and tissues depend, among others, on specific rate of synthesis and degradation of basic energetic compounds. -

The Role of Lysosomal Acid Lipase in Regulation of the Atp
THE ROLE OF LYSOSOMAL ACID LIPASE IN REGULATION OF THE ATP- BINDING CASSETTE TRANSPORTER A1, HIGH DENSITY LIPOPROTEIN AND REVERSE CHOLESTEROL TRANSPORT by Kristin Louise Bowden B.Sc., Queen’s University, 2005 M.Sc., Dalhousie University, 2008. A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in THE FACULTY OF GRADUATE AND POSTDOCTORAL STUDIES (Experimental Medicine) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) October 2013 © Kristin Louise Bowden, 2013. Abstract The key regulator of initial HDL particle formation by cells is the ATP-binding cassette transporter A1 (ABCA1). ABCA1 expression is regulated primarily by oxysterol dependent activation of the liver X receptor (LXR). We investigated the role of lysosomal cholesterol on ABCA1 regulation by studying the lysosomal disorder Cholesteryl Ester Storage Disease (CESD). CESD is caused by genetic mutations in the LIP-A gene that result in only 5% of normal activity of lysosomal acid lipase (LAL), an enzyme that hydrolyzes cholesteryl esters (CE) and triglycerides on internalized lipoproteins specifically within the lysosome. We hypothesized that the flux of unesterified cholesterol out of the lysosomes from LAL- mediated hydrolysis of LDL cholesteryl esters is a key regulator of cellular ABCA1 expression, HDL formation and reverse cholesterol transport (RCT). We found that primary skin fibroblasts derived from individuals with CESD had impaired upregulation of ABCA1 in response to LDL loading, reduced phospholipid and cholesterol efflux to apoA-I, lower production of 27-hydroxycholesterol (27-OH) production in response to LDL loading and reduced α-HDL particle formation. This defect was recapitulated in normal fibroblasts following treatment with LAL inhibitors, whereas, treatment with conditioned medium from normal fibroblasts containing secreted LAL rescued ABCA1 expression, apoA-I-mediated cholesterol efflux, HDL particle formation and production of 27-OH by CESD cells. -

The Physiological Role of Glucocorticoid and Mineralocorticoid Receptor Activation in Zebrafish
University of Calgary PRISM: University of Calgary's Digital Repository Graduate Studies The Vault: Electronic Theses and Dissertations 2019-06-25 The Physiological Role of Glucocorticoid and Mineralocorticoid Receptor Activation in Zebrafish Faught, Leslie Erin Faught, L. E. (2019). The Physiological Role of Glucocorticoid and Mineralocorticoid Receptor Activation in Zebrafish (Unpublished doctoral thesis). University of Calgary, Calgary, AB. http://hdl.handle.net/1880/110575 doctoral thesis University of Calgary graduate students retain copyright ownership and moral rights for their thesis. You may use this material in any way that is permitted by the Copyright Act or through licensing that has been assigned to the document. For uses that are not allowable under copyright legislation or licensing, you are required to seek permission. Downloaded from PRISM: https://prism.ucalgary.ca UNIVERSITY OF CALGARY The Physiological Role of Glucocorticoid and Mineralocorticoid Receptor Activation in Zebrafish by Leslie Erin Faught A THESIS SUBMITTED TO THE FACULTY OF GRADUATE STUDIES IN PARTIAL FULFILMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY GRADUATE PROGRAM IN BIOLOGICAL SCIENCES CALGARY, ALBERTA JUNE, 2019 © Leslie Erin Faught 2019 Abstract Glucocorticoids are key mediators of the vertebrate stress response. In teleosts, the primary glucocorticoid, cortisol, is a ligand for two corticosteroid receptors (CRs), the glucocorticoid receptor (GR) and the mineralocorticoid receptor (MR). The affinity of cortisol for these receptors is markedly different, with MR having an almost 10-fold higher affinity for the ligand compared to GR. This led to the hypothesis 30 years ago in mammals, that MR is responsible for basal cortisol function, while GR is active only when cortisol levels are high. -

The Role of Lipid Metabolism in Aging, Lifespan Regulation, and Age‐Related Disease
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Loughborough University Institutional Repository Received: 6 May 2019 | Revised: 11 August 2019 | Accepted: 4 September 2019 DOI: 10.1111/acel.13048 REVIEW The role of lipid metabolism in aging, lifespan regulation, and age‐related disease Adiv A. Johnson1 | Alexandra Stolzing2,3 1Nikon Instruments, Melville, NY, USA Abstract 2BIOAGE Labs, Richmond, CA, USA 3Loughborough University, Loughborough, An emerging body of data suggests that lipid metabolism has an important role to UK play in the aging process. Indeed, a plethora of dietary, pharmacological, genetic, and Correspondence surgical lipid‐related interventions extend lifespan in nematodes, fruit flies, mice, and Adiv A. Johnson, Nikon Instruments, rats. For example, the impairment of genes involved in ceramide and sphingolipid Melville, NY, USA. Email: [email protected] synthesis extends lifespan in both worms and flies. The overexpression of fatty acid amide hydrolase or lysosomal lipase prolongs life in Caenorhabditis elegans, while the overexpression of diacylglycerol lipase enhances longevity in both C. elegans and Drosophila melanogaster. The surgical removal of adipose tissue extends lifespan in rats, and increased expression of apolipoprotein D enhances survival in both flies and mice. Mouse lifespan can be additionally extended by the genetic deletion of diacyl‐ glycerol acyltransferase 1, treatment with the steroid 17‐α‐estradiol, or a ketogenic diet. Moreover, deletion of the phospholipase A2 receptor improves various health‐ span parameters in a progeria mouse model. Genome‐wide association studies have found several lipid‐related variants to be associated with human aging. For example, the epsilon 2 and epsilon 4 alleles of apolipoprotein E are associated with extreme longevity and late‐onset neurodegenerative disease, respectively. -

Emerging Roles of PHLPP Phosphatases in Cell Signaling
Annual Review of Pharmacology and Toxicology PHLPPing the Script: Emerging Roles of PHLPP Phosphatases in Cell Signaling Timothy R. Baffi,∗ Ksenya Cohen-Katsenelson,∗ and Alexandra C. Newton Department of Pharmacology, University of California, San Diego, La Jolla, California 92093-0721, USA; email: [email protected] Annu. Rev. Pharmacol. Toxicol. 2021. 61:723–43 Keywords First published as a Review in Advance on PHLPP, Akt, PKC, phosphatase, phosphorylation, cancer, transcription, September 30, 2020 inflammation The Annual Review of Pharmacology and Toxicology is online at pharmtox.annualreviews.org Abstract https://doi.org/10.1146/annurev-pharmtox-031820- Whereas protein kinases have been successfully targeted for a variety of dis- 122108 eases, protein phosphatases remain an underutilized therapeutic target, in Copyright © 2021 by Annual Reviews. part because of incomplete characterization of their effects on signaling net- All rights reserved works. The pleckstrin homology domain leucine-rich repeat protein phos- ∗ These authors contributed equally to this article phatase (PHLPP) is a relatively new player in the cell signaling field, and new Access provided by University of California - San Diego on 03/15/21. For personal use only. Annu. Rev. Pharmacol. Toxicol. 2021.61:723-743. Downloaded from www.annualreviews.org roles in controlling the balance among cell survival, proliferation, and apop- tosis are being increasingly identified. Originally characterized for its tumor- suppressive function in deactivating the prosurvival kinase Akt, PHLPP may have an opposing role in promoting survival, as recent evidence suggests. Additionally, identification of the transcription factor STAT1 as a substrate unveils a role for PHLPP as a critical mediator of transcriptional programs in cancer and the inflammatory response. -

MTOR Signaling and Metabolism in Early T Cell Development
G C A T T A C G G C A T genes Review MTOR Signaling and Metabolism in Early T Cell Development Guy Werlen, Ritika Jain and Estela Jacinto * Department of Biochemistry and Molecular Biology, Robert Wood Johnson Medical School, Rutgers University, Piscataway, NJ 08854, USA; [email protected] (G.W.); [email protected] (R.J.) * Correspondence: [email protected]; Tel.: +1-732-235-4476 Abstract: The mechanistic target of rapamycin (mTOR) controls cell fate and responses via its func- tions in regulating metabolism. Its role in controlling immunity was unraveled by early studies on the immunosuppressive properties of rapamycin. Recent studies have provided insights on how metabolic reprogramming and mTOR signaling impact peripheral T cell activation and fate. The contribution of mTOR and metabolism during early T-cell development in the thymus is also emerging and is the subject of this review. Two major T lineages with distinct immune functions and peripheral homing organs diverge during early thymic development; the αβ- and γδ-T cells, which are defined by their respective TCR subunits. Thymic T-regulatory cells, which have immunosup- pressive functions, also develop in the thymus from positively selected αβ-T cells. Here, we review recent findings on how the two mTOR protein complexes, mTORC1 and mTORC2, and the signaling molecules involved in the mTOR pathway are involved in thymocyte differentiation. We discuss emerging views on how metabolic remodeling impacts early T cell development and how this can be mediated via mTOR signaling. Keywords: mTOR; mTORC1; mTORC2; thymocytes; T lymphocytes; early T cell development; T-cell metabolism Citation: Werlen, G.; Jain, R.; Jacinto, E. -

(12) United States Patent (10) Patent No.: US 9,383,367 B1 Liu Et Al
USOO9383367B1 (12) United States Patent (10) Patent No.: US 9,383,367 B1 Liu et al. (45) Date of Patent: Jul. 5, 2016 (54) METHODS OF DETECTING CONJUGATION 6,465,199 B1 10/2002 Craig et al. SITE-SPECIFIC AND HIDDEN 6,762,045 B2 7/2004 Krebs et al. 6,911,335 B2 6/2005 Kapeller-Libermann et al. EPTOPEAANTIGEN 7,022,493 B2 4/2006 Issakani et al. 7,223,556 B1 5/2007 Zhou et al. (76) Inventors: Chunli Liu, Baltimore, MD (US); 7.460,960 B2 12/2008 Lee et al. Bingren Hu, Baltimore, MD (US) 7,491,501 B2 2/2009 Wooten 7,803,553 B2 9/2010 Kojima et al. (*) Notice: Subject to any disclaimer, the term of this 2007, 0037221 A1 2/2007 Blocket al. patent is extended or adjusted under 35 2007/0218069 A1 9, 2007 Gordon et al. U.S.C. 154(b) by 0 days. FOREIGN PATENT DOCUMENTS (22) Filed: Dec. 6, 2011 OTHER PUBLICATIONS Related U.S. Application Data Kirkpatricket al. Quantitative analysis of in vitro ubiquitinated cyclin (60) Provisional application No. 61/420.354, filed on Dec. B1 reveals compex chain topology. Nature Cell Biol. 2006, vol. 8, 7, 2010. No. 7, pp. 700-710 and supporting online material.* Koivunen et al. Principles of immunochemical techniques used in (51) Int. Cl. clinical laboratories. Labmedicine 2006, Vo.37, No. 8, pp. 490-497.* GOIN33/53 (2006.01) Continued CI2O I/34 (2006.01) ( ) CI2O I/37 (2006.01) C07K 16/00 (2006.01) GOIN33/68 (2006.01) Primary Examiner — Shafiqul Haq C07K 6/8 (2006.01) (52) U.SCI2O % 1/61 ( 2006.O1 ) (57) ABSTRACT CPC ............... -

Studies on Triacylglycerol Ester Hydrolase from Bat Adipose Tissue
J. Biosci., Vol.5, Number 1, March 1983, pp. 35–41 © Printed in India. Studies on triacylglycerol ester hydrolase from bat adipose tissue SUBHASH S. PATIL, CHANDA Κ. BHANDARI and VIJAY A. SAWANT Animal Physiology Laboratory, Zoology Department, Shivaji University, Kolhapur 416 004 MS received 20 April 1982; revised 4 October 1982. Abstract. Triacylglycerol ester hydrolase was isolated from bat adipose tissue and characterized. The partially purified enzyme had pH optimum of 8.6 and a Km value of 0.6 mM. The enzyme was denaturated upon freezing and thawing, which was prevented by 25% glycerol. The enzyme was activated by EDTA and NaCl, while it was inhibited by serum and bovine serum albumin. Heparin, sodium fluoride and diisopropyl fluorophosphate had no effect on triacylglycerol ester hydrolase activity. It hydrolyzed triglycerides partially. Triacylglycerol ester hydrolase lost its activity during delipidation but it was reactivated by endogenous lipids and phospholipids, viz. phosphatidyl ethanolamine, phosphatidyl choline and sphingomyelin. The enzyme shows kinetic properties altogether different from lipoprotein lipase and hormone sensitive lipase. Keywords. Bat adipose tissue; triacylglycerol ester hydrolase; bovine serum albumin; phospholipids. Introduction Hormone sensitive lipase [EC 3.1.1.3] and lipoprotein lipase [EC 3.1.1.34] have been extensively studied (Hiromichi and Setsuro, 1974; Mare, 1975; Bolzano, 1977; Setsurp and Yasuyuki, 1977; Anon, 1979). The functional importance of lipase has been shown (Vaughan et al., 1964; Hollenberg, 1965). Matsumura et al. (1976a,b) showed the presence of triglyceride lipase in rat and pig adipose tissue in addition to the occurrence of lipoprotein lipase and hormone sensitive lipase. Triglyceride lipase was active in the absence of serum and was strongly inhibited by bovine serum albumin. -

Biological Chemistry
Ministry of Health of Ukraine Zaporizhzhya State Medical University Biochemistry & Laboratory Diagnostics Department Biological chemistry A manual for independent work at home and in class preparation for licensing examination “KROK 1” on module 1 “General regularities of metabolism. Metabolism of carbohydrates, lipids, amino acids and their regulation” for students of International Faculty (the second year of study) speciality: 7.120 10001 «General Medicine» Zaporizhzhya 2015 ББК 28.072я73 Б63 УДК 577.1(072)=111 Editors: Dr. Hab., professor Aleksandrova K.V. PhD, ass. professor Krisanova N.V. PhD, ass. professor Ivanchenko D.G. PhD, as. profesor Rudko N.P. PhD, assistant Levich S.V. This manual is recommended for II year students of International Faculty of specialty 7.12010001 "General medicine" studying biological chemistry, as additional material to prepare for practical training module 1 and licensing exam “KROK 1: General medical training”. Reviewers: • Head of Biological Chemistry Department of National University of Pharmacy, doctor of biological science, professor Zagayko A.L. • Professor of Chemistry Department of Zaporizhzhya National University, doctor of pharmaceutical science, professor Omelianchyk L.O. ББК 28.072я73 УДК 577.1(072)=111 © Aleksandrova K.V., Krisanova N.V., Ivanchenko D.G., Rudko N.P., Levich S.V., 2015 1 CONTENT Introduction…………………………………………………………………... 3 Classification, physicochemical properties and functions of simple proteins in humans. The methods for indication, separation and release of proteins from biological fluids. Created by Levich S.V..……....................................... 4 Conjugated proteins. The methods of allocation and quantitative determination of proteins in biological fluids. Created by Levich S.V..... 18 Enzymes: Structure and physicochemical properties. classification and nomenclature of enzymes. -

Genomics and Proteomics of Vertebrate Cholesterol Ester Lipase (LIPA) and Cholesterol 25-Hydroxylase (CH25H)
3 Biotech (2011) 1:99–109 DOI 10.1007/s13205-011-0013-9 ORIGINAL ARTICLE Genomics and proteomics of vertebrate cholesterol ester lipase (LIPA) and cholesterol 25-hydroxylase (CH25H) Roger S. Holmes • John L. VandeBerg • Laura A. Cox Received: 24 March 2011 / Accepted: 31 May 2011 / Published online: 3 August 2011 Ó The Author(s) 2011. This article is published with open access at Springerlink.com Abstract Cholesterol ester lipase (LIPA; EC 3.1.1.13) coding exons, while all vertebrate CH25H genes were and cholesterol 25-hydroxylase (CH25H; EC 1.14.99.48) without introns. Phylogenetic analysis demonstrated the play essential role in cholesterol metabolism in the body by distinct nature of the vertebrate LIPA gene and protein hydrolysing cholesteryl esters and triglycerides within family in comparison with other vertebrate acid lipases and lysosomes (LIPA) and catalysing the formation of has apparently evolved from an ancestral LIPA gene which 25-hydroxycholesterol from cholesterol (CH25H) which predated the appearance of vertebrates. acts to repress cholesterol biosynthesis. Bioinformatic methods were used to predict the amino acid sequences, Keywords Vertebrates Á Lipase A Á Cholesterol structures and genomic features of several vertebrate LIPA 25-hydroxylase Á Cholesterol metabolism and CH25H genes and proteins, and to examine the phy- logeny of vertebrate LIPA. Amino acid sequence align- ments and predicted subunit structures enabled the Introduction identification of key sequences previously reported for human LIPA and CH25H and transmembrane