United States Patent Office Patented Mar
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Full Text in Pdf Format
DISEASES OF AQUATIC ORGANISMS Published July 30 Dis Aquat Org Oral pharmacological treatments for parasitic diseases of rainbow trout Oncorhynchus mykiss. 11: Gyrodactylus sp. J. L. Tojo*, M. T. Santamarina Department of Microbiology and Parasitology, Laboratory of Parasitology, Faculty of Pharmacy, Universidad de Santiago de Compostela, E-15706 Santiago de Compostela, Spain ABSTRACT: A total of 24 drugs were evaluated as regards their efficacy for oral treatment of gyro- dactylosis in rainbow trout Oncorhj~nchusmykiss. In preliminary trials, all drugs were supplied to infected fish at 40 g per kg of feed for 10 d. Twenty-two of the drugs tested (aminosidine, amprolium, benznidazole, b~thionol,chloroquine, diethylcarbamazine, flubendazole, levamisole, mebendazole, n~etronidazole,mclosamide, nitroxynil, oxibendazole, parbendazole, piperazine, praziquantel, roni- dazole, secnidazole, tetramisole, thiophanate, toltrazuril and trichlorfon) were ineffective Triclabenda- zole and nitroscanate completely eliminated the infection. Triclabendazole was effective only at the screening dosage (40 g per kg of feed for 10 d), while nitroscanate was effective at dosages as low as 0.6 g per kg of feed for 1 d. KEY WORDS: Gyrodactylosis . Rainbow trout Treatment. Drugs INTRODUCTION to the hooks of the opisthohaptor or to ulceration as a result of feeding by the parasite. The latter is the most The monogenean genus Gyrodactylus is widespread, serious. though some individual species have a restricted distri- Transmission takes place largely as a result of direct bution. Gyrodactyloses affect numerous freshwater contact between live fishes, though other pathways species including salmonids, cyprinids and ornamen- (contact between a live fish and a dead fish, or with tal fishes, as well as marine fishes including gadids, free-living parasites present in the substrate, or with pleuronectids and gobiids. -

Development and Optimisation of a Multi-Residue Method for The
Supplementary Material Development and optimisation of a multi-residue method for the determination of 40 anthelmintic compounds in environmental water samples by solid phase extraction (SPE) with LC-MS/MS detection. Damien Mooney 1, 4, 5, *, Catherine Coxon 1, 5, Karl G Richards 2, 5, Laurence Gill 3, 5, Per-Erik Mellander 2 and Martin Danaher 4 1 School of Natural Sciences, Geology Department, Trinity College Dublin, Ireland; [email protected] and [email protected] 2 Environment, Soils and Land-Use Department, Environment Research Centre, Teagasc, Johnstown Castle, Wexford, Ireland; [email protected] and [email protected] 3 Department of Civil, Structural and Environmental Engineering, Trinity College Dublin, Ireland; [email protected] 4 Food Safety Department, Teagasc Food Research Centre, Ashtown, Dublin 15, Ireland; [email protected] and [email protected] 5 Groundwater spoke, Irish Centre for Research in Applied Geosciences (iCRAG), Ireland * Correspondence: [email protected] (DM) or [email protected] (MD); Tel.: +353-1-8059500 Received: date; Accepted: date; Published: date Contents Figure S1. Structures of anthelmintic compounds by structural class • (a) structures of benzimidazole anthelmintics • (b) structures of macrocyclic lactone anthelmintics • (c) structures of organophosphate anthelmintics • (d) structures of salicylanilide and substituted phenol anthelmintics • (e) structures of amino-acetonitrile derivative anthelmintics • (f) structures of tetrahydropyrimidines (MOR) and imidazothiazole (LEV) anthelmintics • (g) structure of one miscellaneous anthelmintic (CLOR) Figure S2. Mean recovery and precision (%RSD, presented as error bars) for assessment of sorbent mass (200 mg vs. 500 mg) each using three elution volume (10, 15 and 20.mL) Figure S3 (a). -

Recommended Classification of Pesticides by Hazard and Guidelines to Classification 2019 Theinternational Programme on Chemical Safety (IPCS) Was Established in 1980
The WHO Recommended Classi cation of Pesticides by Hazard and Guidelines to Classi cation 2019 cation Hazard of Pesticides by and Guidelines to Classi The WHO Recommended Classi The WHO Recommended Classi cation of Pesticides by Hazard and Guidelines to Classi cation 2019 The WHO Recommended Classification of Pesticides by Hazard and Guidelines to Classification 2019 TheInternational Programme on Chemical Safety (IPCS) was established in 1980. The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals. This publication was developed in the IOMC context. The contents do not necessarily reflect the views or stated policies of individual IOMC Participating Organizations. The Inter-Organization Programme for the Sound Management of Chemicals (IOMC) was established in 1995 following recommendations made by the 1992 UN Conference on Environment and Development to strengthen cooperation and increase international coordination in the field of chemical safety. The Participating Organizations are: FAO, ILO, UNDP, UNEP, UNIDO, UNITAR, WHO, World Bank and OECD. The purpose of the IOMC is to promote coordination of the policies and activities pursued by the Participating Organizations, jointly or separately, to achieve the sound management of chemicals in relation to human health and the environment. WHO recommended classification of pesticides by hazard and guidelines to classification, 2019 edition ISBN 978-92-4-000566-2 (electronic version) ISBN 978-92-4-000567-9 (print version) ISSN 1684-1042 © World Health Organization 2020 Some rights reserved. -
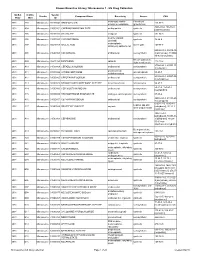
Known Bioactive Library: Microsource 1 - US Drug Collection
Known Bioactive Library: Microsource 1 - US Drug Collection ICCB-L ICCB-L Vendor Vendor Compound Name Bioactivity Source CAS Plate Well ID antifungal, inhibits Penicillium 2091 A03 Microsource 00200046 GRISEOFULVIN 126-07-8 mitosis in metaphase griseofulvum 3505-38-2, 486-16-8 2091 A04 Microsource 01500161 CARBINOXAMINE MALEATE antihistaminic synthetic [carbinoxamine] 2091 A05 Microsource 00200331 SALSALATE analgesic synthetic 552-94-3 muscle relaxant 2091 A06 Microsource 01500162 CARISOPRODOL synthetic 78-44-4 (skeletal) antineoplastic, 2091 A07 Microsource 00210369 GALLIC ACID insect galls 149-91-7 astringent, antibacterial 66592-87-8, 50370-12- 2091 A08 Microsource 01500163 CEFADROXIL antibacterial semisynthetic 2 [anhydrous], 119922- 89-9 [hemihydrate] Rheum palmatum, 2091 A09 Microsource 00211468 DANTHRON cathartic 117-10-2 Xyris semifuscata 27164-46-1, 25953-19- 2091 A10 Microsource 01500164 CEFAZOLIN SODIUM antibacterial semisynthetic 9 [cefazolin] glucocorticoid, 2091 A11 Microsource 00300024 HYDROCORTISONE adrenal glands 50-23-7 antiinflammatory 64485-93-4, 63527-52- 2091 A12 Microsource 01500165 CEFOTAXIME SODIUM antibacterial semisynthetic 6 [cefotaxime] 2091 A13 Microsource 00300029 DESOXYCORTICOSTERONE ACETATE mineralocorticoid adrenocortex 56-47-3 58-71-9, 153-61-7 2091 A14 Microsource 01500166 CEPHALOTHIN SODIUM antibacterial semisynthetic [cephalothin] 2091 A15 Microsource 00300034 TESTOSTERONE PROPIONATE androgen, antineoplastic semisynthetic 57-85-2 24356-60-3, 21593-23- 2091 A16 Microsource 01500167 CEPHAPIRIN SODIUM -

Drench Resistance Status Type of Worm Barber's Pole Scour Worms Nodule Liverfluke Useful Information Summer Rainfall Other
Drench Resistance Status Type of Worm Barber’s pole Scour Nodule Liverfluke Useful information worms Summer Other Rainfall regions Benzimidazole (BZ) Yes*** Yes** Yes*** Yes* BZs may still be useful against roundworms when administered with unrelated actives in combination products or in a long-acting product (capsule) when given with an effective primer Levamisole / morantel (LEV) Yes** Yes*** Levamisole may still be useful when administered with unrelated actives in combination products. Organophosphate (OP) Yes* Yes* Yes* - Salicylanilides/Phenols (SAL-P) Yes*** Yes* - - Yes* Macrocyclic lactone (ML) Yes*** Yes** Yes* Unless resistance is severe, MLs may still be useful when administered with unrelated actives in combination products. Moxidectin-based long-acting products may still be useful when given with an effective primer Amino-acetonitrile derivative (AAD) Unknown Unknown Unknown - Unknown Triclabendazole (BZ Flukicide) - - - - Yes* Isoquinolone (ISO) Benzimidazole and levamisole (BZ and LEV) Yes** Unknown Yes*** Yes* Unless resistance is severe, these combinations may still be useful when administered with unrelated actives in triple or quadruple combination products. Activity of the BZ group also aids in control of adult liver fluke. Benzimidazole and salicylanilide (BZ and SAL-P) Yes*** Yes*** Benzimidazole and triclabendazole (BZ and BZ Yes*** Yes*** Unknown Flukicide) Macrocyclic lactone and salicylanilide (ML and SAL-P) Yes*** Yes*/** Macrocyclic lactone and triclabendazole (ML and BZ Yes*** Yes** Yes*/** Yes* Flukicide) Macrocyclic lactone and isoquinolone (ML and ISO) See isoquinolone and macrocyclic lactone for resistance notes Benzimidazole and macrocyclic lactone and Yes*/** Yes*/** Yes* levamisole (BZ and ML and LEV) Benzimidazole and organophosphate and (BZ and Yes* Yes* Because OPs have variable activity against scour worms and immature barber’s pole worm, the OP) efficacy of this combination against these worms also depends on the resistance status of benzimidazole (see benzimidazole for resistance note). -

2.Anthelmintics
Anthelmintic Anthelmintic or antihelminthics are a group of antiparasitic drugs that expel parasitic worms (helminths) and other internal parasites from the body by either stunning or killing them and without causing significant damage to the host. They may also be called vermifuges (those that stun) or vermicides (those that kill). Anthelmintics are used to treat people who are infected by helminths, a condition called helminthiasis. These drugs are also used to treat infected animals. Antiparasitics that specifically target worms of the genus Ascaris are called ascaricides. Classification: Benzimidazoles: Albendazole – effective against threadworms, roundworms, whipworms, tapeworms, hookworms Mebendazole – effective against various nematodes Thiabendazole – effective against various nematodes Fenbendazole – effective against various parasites Triclabendazole – effective against liver flukes Flubendazole – effective against most intestinal parasites Abamectin (and by extension ivermectin) - effective against most common intestinal worms, except tapeworms, for which praziquantel is commonly used in conjunction for mass dewormings Diethylcarbamazine – effective against Wuchereria bancrofti, Brugia malayi, Brugia timori, and Loa loa. Pyrantel pamoate – effective against most nematode infections residing within the intestines `Levamisole Salicylanilide – mitochondrial un-couplers (used only for flatworm infections): Niclosamide Oxyclozanide Nitazoxanide – readily kills Ascaris lumbricoides,[5] and also possess antiprotozoal effects[6] -

P-Glycoprotein Selection in Strains of Haemonchus Contortus Resistant to Benzimidazoles William J
Available online at www.sciencedirect.com Veterinary Parasitology 152 (2008) 101–107 www.elsevier.com/locate/vetpar P-glycoprotein selection in strains of Haemonchus contortus resistant to benzimidazoles William J. Blackhall *, Roger K. Prichard, Robin N. Beech Institute of Parasitology, McGill University, 21111 Lakeshore Road, Ste.-Anne-de-Bellevue, QC H9X 3V4, Canada Received 3 January 2007; received in revised form 28 November 2007; accepted 4 December 2007 Abstract Anthelmintic resistance in parasitic nematodes of livestock is a chronic problem in many parts of the world. Benzimidazoles are effective, broad-spectrum anthelmintics that bind to and selectively depolymerise microtubules. Resistance to the benzimidazoles, however, developed quickly and is caused by genetic changes in genes encoding b-tubulins, subunits of microtubules. In Haemonchus contortus, resistance to avermectins has been correlated with genetic changes at a gene encoding a P-glycoprotein, a cell membrane transport protein that has a very high affinity for ivermectin. The substrate specificity of P-glycoprotein is very broad, and resistance to benzimidazoles can be modulated by lectins specific for P-glycoprotein. We investigated the possibility that genetic changes in P-glycoprotein might be correlated with benzimidazole resistance in nematodes. An analysis of restriction fragment length polymorphisms of a P-glycoprotein gene from a sensitive and a cambendazole-selected strain of H. contortus, derived from the sensitive strain, showed a significant difference in allele frequencies between strains. The frequency of one allele in particular increased substantially. The same allele was also found at a high frequency in an independently derived thiabendazole- selected field isolate. We present genetic evidence of selection at a P-glycoprotein locus during selection for benzimidzole resistance in H. -

Niclosamide Is Active in Vitro Against Mycetoma Pathogens
molecules Article Niclosamide Is Active In Vitro against Mycetoma Pathogens Abdelhalim B. Mahmoud 1,2,3,† , Shereen Abd Algaffar 4,† , Wendy van de Sande 5, Sami Khalid 4 , Marcel Kaiser 1,2 and Pascal Mäser 1,2,* 1 Department of Medical Parasitology and Infection Biology, Swiss Tropical and Public Health Institute, 4051 Basel, Switzerland; [email protected] (A.B.M.); [email protected] (M.K.) 2 Faculty of Science, University of Basel, 4001 Basel, Switzerland 3 Faculty of Pharmacy, University of Khartoum, Khartoum 11111, Sudan 4 Faculty of Pharmacy, University of Science and Technology, Omdurman 14411, Sudan; [email protected] (S.A.A.); [email protected] (S.K.) 5 Erasmus Medical Center, Department of Medical Microbiology and Infectious Diseases, 3000 Rotterdam, The Netherlands; [email protected] * Correspondence: [email protected]; Tel.: +41-61-284-8338 † These authors contributed equally to this work. Abstract: Redox-active drugs are the mainstay of parasite chemotherapy. To assess their repurposing potential for eumycetoma, we have tested a set of nitroheterocycles and peroxides in vitro against two isolates of Madurella mycetomatis, the main causative agent of eumycetoma in Sudan. All the tested compounds were inactive except for niclosamide, which had minimal inhibitory concentrations of around 1 µg/mL. Further tests with niclosamide and niclosamide ethanolamine demonstrated in vitro activity not only against M. mycetomatis but also against Actinomadura spp., causative agents of actinomycetoma, with minimal inhibitory concentrations below 1 µg/mL. The experimental compound MMV665807, a related salicylanilide without a nitro group, was as active as niclosamide, indicating that the antimycetomal action of niclosamide is independent of its redox chemistry (which Citation: Mahmoud, A.B.; Abd Algaffar, S.; van de Sande, W.; Khalid, is in agreement with the complete lack of activity in all other nitroheterocyclic drugs tested). -

Activity of Levamisole, Pyrantel Tartrate, and Rafoxanide Against
OF WASHINGTON, VOLUME 38, NUMBER 2, JULY 1971 203 Activity of Levamisole, Pyrantel Tartrate, and Rafoxanide Against Two Thiabendazole-tolerant Isolates of Haemonchus contortus, and Two Species of Trichostrongylus, in Sheep M. L. COLGLAZIER, K. C. KATES, AND F. D. ENZIE National Animal Parasite Laboratory, Veterinary Sciences Research Division, ARS, USDA, Beltsville, Maryland 20705 ABSTRACT: The activity of 3 of the newer anthelmintics against two local thiabendazole-tolerant isolates of Haemonchus contortus (BPL-2 and AH-2), as well as against Trichostrongylus axei and T. colubri- formis, was compared in experimentally infected lambs, using the method of the controlled anthelmintic test. Twenty of the 40 lambs on test were each given per os 5,000 infective larvae of the BPL-2 isolate of H. contortus, and the other 20 were given 5,000 infective larvae of the AH-2 isolate. Also, each lamb was given 18,000 T. axei and 18,000 T. colubriformis infective larvae. Single doses of the test drugs were given to the appropriate groups of lambs 21 clays postinfection, and all lambs were killed for worm counts 6 to 7 days later. Levamisole was given at 8ing/kg of body weight, pyrantel tartrate at 25 mg/kg, and rafoxanide at 5 mg/kg. All 3 anthelmintics were markedly effective (99-100%) against the 2 isolates of H. contortus. Levamisole was very effective also (95-99%) against the 2 species of Trichostrongylus. Pyrantel tartrate was highly effective against T. axei (99%), but substantially less ef- fective (67%) against T. colubriformis. Rafoxanide showed no activity against Trichostrongylus spp. -

ZOLVIX, INN: Monepantel
14 July 2011 EMA/161969/2012 EPAR Scientific Discussion post-authorisation update for ZOLVIX International Non-proprietary Name: Monepantel EU/2/09/101/001-010 Scope: Extension of the indications to H. contortus strains resistant to salicylanilides. 7 Westferry Circus ● Canary Wharf ● London E14 4HB ● United Kingdom Telephone +44 (0)20 7418 8400 Facsimile +44 (0)20 7418 8447 E-mail [email protected] Website www.ema.europa.eu An agency of the European Union © European Medicines Agency, 2012. Reproduction is authorised provided the source is acknowledged. 1. Background information on the variation 1.1. Submission of the variation application In accordance with Article 16 of Commission Regulation (EC) No. 1234/2008, the marketing authorisation holder, Novartis Healthcare A/S, submitted to the European Medicines Agency (the Agency) on 20 December 2010 an application for a type II variation for ZOLVIX. 1.2. Scope of the variation Previous Proposed SPC SPC 4.2 Indications for use, specifying the target 4.2 Indications for use, specifying the species target species The veterinary medicinal product is effective against The veterinary medicinal product is effective strains of these parasites resistant to against strains of these parasites resistant to (pro)benzimidazoles, levamisole, morantel and (pro)benzimidazoles, levamisole, morantel, macrocyclic lactones. macrocyclic lactones and salicylanilides. PACKAGE LEAFLET PACKAGE LEAFLET 4. INDICATION 4. INDICATION The veterinary medicinal product is effective against The veterinary medicinal product is effective strains of these parasites resistant to against strains of these parasites resistant to (pro)benzimidazoles, levamisole, morantel and (pro)benzimidazoles, levamisole, morantel, macrocyclic lactones. macrocyclic lactones and salicylanilides. -

Pharmacophore DESIGN and PHARMACOCHEMICAL DEVELOPMENT of NEW ANTHELMINTICS with BENZIMIDAZOLYL ARYLPROPENONE PROFILE: STRUCTURE
Pharmacophore, 11(1) 2020, Pages: 58-66 Pharmacophore ISSN-2229-5402 Journal home page: http://www.pharmacophorejournal.com DESIGN AND PHARMACOCHEMICAL DEVELOPMENT OF NEW ANTHELMINTICS WITH BENZIMIDAZOLYL ARYLPROPENONE PROFILE: STRUCTURE-ACTIVITY RELATIONSHIP Ouattara Mahama1*, N'Guessan Deto Jean-Paul1, Coulibaly Songuigama1, Sissouma Drissa2, Koné W Mamidou 3 1. Department of Therapeutic Chemistry and Organic Chemistry, University of Félix Houphouët-Boigny, 01 BP V34 Abidjan, Ivory Coast 2. Laboratory of Organic Chemistry and Natural Substances,, University of Félix Houphouët-Boigny, 01 BP V34 Abidjan, Ivory Coast 3. Swiss Center for Scientific Research in Ivory Coast, 01 BP 1303 Abidjan. ARTICLE INFO A B S T R A C T Received: On the basis of our previous study, which had demonstrated the strong anthelmintic potential of 08 Aug 2019 benzimidazole-2-arylpropenones, we proposed to extend the anthelmintic evaluation by testing anovel series of benzimidazole against Haemonchus contortus. The aimof this work is to appreciate Received in revised form: the impact of the bioisoteric replacement of arylpropenone by arylacrylonitrile.Furthermore, we 14 Jan 2020 have established the structural elements necessary to get good anthelmintic activities in series of 2- Accepted: substituted benzimidazoles. The structure-activity relationship studies made after all biological 22 Jan 2020 evaluations gathered, revealed that in series benzimidazolyl-phenylpropenones, excellent Available online: anthelmintic activities similar to those of Ivermectin have been achieved with the unsubstituted 28 Feb 2020 phenyl derivative, with the metahydroxyphenyl derivative, and by replacing the benzene homocycle with a heterocycle like pyridine or furan. Of all the variations made around the benzimidazole heterocycle, it appears that the introduction of a chlorine atom on one of the potential sites of metabolism of benzimidazole (C-5), coupled with the non-substitution of pyrrolic nitrogen, is Keywords: Benzimidazole. -

I Scolicidal Effect of Salicylanilide and Bisphenol Derivatives Against Ech1nococcus Granulosus in Vitro
STUDIES ON EFFECTS OF DRUGS UPON PROTOSCOLECES OF ECHINOCOCCUS GRANULOSUS IN VITRO Title : I SCOLICIDAL EFFECT OF SALICYLANILIDE AND BISPHENOL DERIVATIVES AGAINST ECH1NOCOCCUS GRANULOSUS IN VITRO Author(s) SAKAMOTO, Tsukasa; GEMMELL, Michael A. Citation Japanese Journal of Veterinary Research, 23(3), 81-94 Issue Date 1975-07 DOI 10.14943/jjvr.23.3.81 Doc URL http://hdl.handle.net/2115/2067 Type bulletin (article) File Information KJ00002371208.pdf Instructions for use Hokkaido University Collection of Scholarly and Academic Papers : HUSCAP Jap. J. "vet. Res., 23, 81-94 (1975) STUDIES ON EFFECTS OF DRUGS UPON PROTOSCOLECES OF ECHI~;OCOCCUS ORANULOSUS IN VITRO I SCOLICIDAL EFFECT OF SALICYLANILIDE AND BISPHENOL DERIV ATIVES AGAINST ECHINOCOCCUS GRANULOSUS IN VITRO*l Tsukasa SAKAMOTO and Michael A. GEMMELL *2 Department of Parasitolog)' Faculty of Veterinary lvledicine Hokkaido Uni'eersity, Sapporo, Japan (Received for publication, February 10, 1975) The protoscoleces of Echinococcus granulosus (BATSCH, 1786) were incubated for ten days in media to which a range of salicylanilide- and bisphenol-derivatives and other compounds were added at the rates of 1, 10 and 100 r per ml. The main culture medium was 20 % inactivated calf serum with a 1: 1 mixture of medium 199 and HANKS' solution containing 05 ero lactalbumin hydrolysate and 0.1 % yeast extract. The survival rates of the treated and control protoscoleces were compared. Generally, halogenated salicylanilide derivatives showed the highest scolicidal effect and halogenated derivatives of diphenyl sulfide and diphenylmethane ranked next in lethal effect. The intensity of the scolicidal action of salicylanilide deriv atives increased with the addition of some halogen atoms.