Rewiring of the Ftsh Regulatory Network by a Single Nucleotide
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Product Information
Product Information Betaine Aldehyde (chloride) Item No. 17270 CAS Registry No.: 7758-31-8 Formal Name: N,N,N-trimethyl-2-oxo- ethanaminium, monochloride MF: C5H12NO • Cl H FW: 137.6 • Cl- Purity: N+ ≥95% O Stability: ≥2 years at -20°C Supplied as: A crystalline solid Laboratory Procedures For long term storage, we suggest that betaine aldehyde (chloride) be stored as supplied at -20°C. It should be stable for at least two years. Betaine aldehyde (chloride) is supplied as a crystalline solid. A stock solution may be made by dissolving the betaine aldehyde (chloride) in the solvent of choice. Betaine aldehyde (chloride) is soluble in the organic solvents DMSO, which should be purged with an inert gas, at a concentration of approximately 1 mg/ml. Further dilutions of the stock solution into aqueous buffers or isotonic saline should be made prior to performing biological experiments. Ensure that the residual amount of organic solvent is insignificant, since organic solvents may have physiological effects at low concentrations. Organic solvent-free aqueous solutions of betaine aldehyde (chloride) can be prepared by directly dissolving the crystalline solid in aqueous buffers. The solubility of betaine aldehyde (chloride) in PBS, pH 7.2, is approximately 5 mg/ml. We do not recommend storing the aqueous solution for more than one day. Betaine aldehyde is the physiological intermediate in the oxidation of choline to betaine. This step is involved in glycine, serine, and threonine metabolism. Related Products For a list of related products please visit: www.caymanchem.com/catalog/17270 Cayman Chemical Mailing address 1180 E. -

Discovery of a Choline-Responsive Transcriptional Regulator in Burkholderia Xenovorans
Journal of Molecular Biology Research; Vol. 3, No. 1; 2013 ISSN 1925-430X E-ISSN 1925-4318 Published by Canadian Center of Science and Education Discovery of a Choline-Responsive Transcriptional Regulator in Burkholderia xenovorans Ricardo Martí-Arbona1, Tuhin S. Maity1, John M. Dunbar1, Clifford J. Unkefer1 & Pat J. Unkefer1 1 Los Alamos National Laboratory, Los Alamos, NM 87545, United States Correspondence: Ricardo Martí-Arbona, Los Alamos National Laboratory, Los Alamos, NM 87545, United States. E-mail: [email protected] Received: August 8, 2013 Accepted: August 20, 2013 Online Published: August 22, 2013 doi:10.5539/jmbr.v3n1p91 URL: http://dx.doi.org/10.5539/jmbr.v3n1p91 Abstract The search for effectors of novel transcriptional regulators is a challenging task. Here, we present the prediction and validation of an effector for a novel transcriptional regulator (TR). The clustering of genes around the gene coding for Bxe_A0425, a TR in Burkholderia xenovorans LB400 and its closest orthologs, suggests the conservation of a functional operon composed a several open reading frames from which a TR, a transporter, and two oxidoreductases can be easily identified. A search of operons containing these functional components revealed a remarkable resemblance of this system to the evolutionarily convergent and functionally conserved operons found in Escherichia coli, Bacillus subtilis, Staphylococcus xylosus and Pseudomonas aeruginosa. These operons are involved in the uptake and catabolism of choline to create the potent osmo-protectant molecule glycine betaine. We used frontal affinity chromatography coupled to mass spectrometry to screen for the binding of choline and other intermediates of the glycine biosynthesis pathway to the TR Bxe_0425. -

Metabolism and Occurrence of Methanogenic and Sulfate-Reducing Syntrophic Acetate Oxidizing Communities in Haloalkaline Environments
Delft University of Technology Metabolism and occurrence of methanogenic and sulfate-reducing syntrophic acetate oxidizing communities in haloalkaline environments Timmers, Peer H.A.; Vavourakis, Charlotte D.; Kleerebezem, Robbert; Sinninghe Damsté, Jaap S.; Muyzer, Gerard; Stams, Alfons J.M.; Sorokin, Dimity Y.; Plugge, Caroline M. DOI 10.3389/fmicb.2018.03039 Publication date 2018 Document Version Final published version Published in Frontiers in Microbiology Citation (APA) Timmers, P. H. A., Vavourakis, C. D., Kleerebezem, R., Sinninghe Damsté, J. S., Muyzer, G., Stams, A. J. M., Sorokin, D. Y., & Plugge, C. M. (2018). Metabolism and occurrence of methanogenic and sulfate- reducing syntrophic acetate oxidizing communities in haloalkaline environments. Frontiers in Microbiology, 9(DEC), [3039]. https://doi.org/10.3389/fmicb.2018.03039 Important note To cite this publication, please use the final published version (if applicable). Please check the document version above. Copyright Other than for strictly personal use, it is not permitted to download, forward or distribute the text or part of it, without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license such as Creative Commons. Takedown policy Please contact us and provide details if you believe this document breaches copyrights. We will remove access to the work immediately and investigate your claim. This work is downloaded from Delft University of Technology. For technical reasons the number of authors shown on this cover page is limited to a maximum of 10. fmicb-09-03039 December 6, 2018 Time: 15:4 # 1 ORIGINAL RESEARCH published: 10 December 2018 doi: 10.3389/fmicb.2018.03039 Metabolism and Occurrence of Methanogenic and Sulfate-Reducing Syntrophic Acetate Oxidizing Communities in Haloalkaline Environments Peer H. -

Viewed by Maurel, 1997)
CLONING AND CHARACTERIZATION OF GENES RELATED TO BETAINE SYNTHESIS, THE EFFECT OF SALT ON CELL DEATH, AND COMPETITION ON ATRIPLEX PROSTRATA A dissertation presented to the faculty of the College of Arts and Sciences of Ohio University In partial fulfillment of the requirements for the degree Doctor of Philosophy Li-Wen Wang August 2002 This dissertation entitled CLONING AND CHARACTERIZATION OF GENES RELATED TO BETAINE SYNTHESIS, THE EFFECT OF SALT ON CELL DEATH, AND COMPETITION ON ATRIPLEX PROSTRATA BY LI-WEN WANG has been approved for the Department of Environmental and Plant Biology and the College of Arts and Sciences by Allan M. Showalter Professor of Environmental and Plant Biology Leslie A. Flemming Dean, College of Arts and Sciences WANG, LI-WEN Ph.D. August 2002. Environmental and Plant Biology / Molecular and Cellular Biology Cloning and Characterization of Genes Related to Betaine, the Effect of Salt on Cell Death and Competition on Atriplex Prostrata (248 pp.) Director of Dissertation: Allan M. Showalter Soil salinity is a major concern to agriculture all over the world because excess salts in the soil inhibit crop growth. Halophytes such as Atriplex prostrata are able to grow and reproduce in saline environments. One of the reasons A. prostrata is salt- tolerant is that it accumulates osmoprotectants such as glycine betaine in the cytosol and sequesters Na+ and Cl- into the vacuoles. In higher plants, glycine betaine is synthesized via the two-step oxidation of choline catalyzed by choline monooxygenase (CMO) and betaine aldehyde dehydrogenase (BADH). The cDNAs encoding CMO and BADH are cloned from A. -

Structure-Guided Function Discovery of an NRPS-Like Glycine Betaine Reductase for Choline Biosynthesis in Fungi
Structure-guided function discovery of an NRPS-like glycine betaine reductase for choline biosynthesis in fungi Yang Hai (海洋)a, Arthur M. Huangb, and Yi Tanga,b,1 aDepartment of Chemical and Biomolecular Engineering, University of California, Los Angeles, CA 90095; and bDepartment of Chemistry and Biochemistry, University of California, Los Angeles, CA 90095 Edited by Wilfred A. van der Donk, Howard Hughes Medical Institute and University of Illinois, Urbana–Champaign, Urbana, IL, and accepted by Editorial Board Member Stephen J. Benkovic April 10, 2019 (received for review February 27, 2019) Nonribosomal peptide synthetases (NRPSs) and NRPS-like enzymes focused on a fungal CAR-like protein with an unknown function. It have diverse functions in primary and secondary metabolisms. By is distinguished from all other CARs due to an extra C-terminal using a structure-guided approach, we uncovered the function of a YdfG-like short-chain dehydrogenase/reductase domain. We NRPS-like enzyme with unusual domain architecture, catalyzing named this protein ATRR and the corresponding gene atrr after its two sequential two-electron reductions of glycine betaine to choline. unusual domain architecture (A-T-R1-R2). The presence of two Structural analysis based on the homology model suggests cation-π fused R domains in ATRR implies that it could catalyze two con- interactions as the major substrate specificity determinant, which was secutive two-electron reductions of a carboxylic acid to yield an verified using substrate analogs and inhibitors. Bioinformatic analysis alcohol (Fig. 1B). Moreover, a genomic survey reveals that atrr indicates this NRPS-like glycine betaine reductase is highly conserved genes are widespread but exclusive to eukaryotes, mostly in fungi and widespread in kingdom fungi. -

Synthesis of the Osmoprotectant Glycine Betaine in Bacillus Subtilis: Characterization of the Gbsab Genes
JOURNAL OF BACTERIOLOGY, Sept. 1996, p. 5121–5129 Vol. 178, No. 17 0021-9193/96/$04.0010 Copyright q 1996, American Society for Microbiology Synthesis of the Osmoprotectant Glycine Betaine in Bacillus subtilis: Characterization of the gbsAB Genes 1,2 1,2 3 1,2 JENS BOCH, BETTINA KEMPF, ROLAND SCHMID, AND ERHARD BREMER * Max-Planck Institute for Terrestrial Microbiology1 and Laboratory for Microbiology, University of Marburg,2 Marburg, and Laboratory for Microbiology, University of Osnabru¨ck, Osnabru¨ck,3 Federal Republic of Germany Received 12 February 1996/Accepted 19 June 1996 Synthesis of the osmoprotectant glycine betaine from the exogenously provided precursor choline or glycine betaine aldehyde confers considerable osmotic stress tolerance to Bacillus subtilis in high-osmolarity media. Using an Escherichia coli mutant (betBA) defective in the glycine betaine synthesis enzymes, we cloned by functional complementation the genes that are required for the synthesis of the osmoprotectant glycine betaine in B. subtilis. The DNA sequence of a 4.1-kb segment from the cloned chromosomal B. subtilis DNA was established, and two genes (gbsA and gbsB) whose products were essential for glycine betaine biosynthesis and osmoprotection were identified. The gbsA and gbsB genes are transcribed in the same direction, are separated by a short intergenic region, and are likely to form an operon. The deduced gbsA gene product exhibits strong sequence identity with members of a superfamily of specialized and nonspecialized aldehyde dehydrogenases. This superfamily comprises glycine betaine aldehyde dehydrogenases from bacteria and plants with known involvement in the cellular adaptation to high-osmolarity stress and drought. The deduced gbsB gene product shows significant similarity to the family of type III alcohol dehydrogenases. -

Structure-Guided Function Discovery of an NRPS-Like Glycine Beta
bioRxiv preprint doi: https://doi.org/10.1101/559534; this version posted February 24, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Classification BIOLOGICAL SCIENCES: Biochemistry Structure-guided function discovery of an NRPS-like glycine beta- ine reductase for choline biosynthesis in fungi Yang Hai1, Arthur Huang2, Yi Tang1,2* 1Department of Chemical and Biomolecular Engineering, 2Department of Chemistry and Bio- chemistry, University of California, Los Angeles, California 90095, United States * Email: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/559534; this version posted February 24, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. ABSTRACT Nonribosomal peptide synthetases (NRPS) and NRPS-like enzymes have diverse func- tions in primary and secondary metabolism. By using a structure-guided approach, we uncov- ered the function of an NRPS-like enzyme with unusual domain architecture, catalyzing two sequential two-electron reductions of glycine betaine to choline. Structural analysis based on homology model suggests cation-p interactions as the major substrate specificity determinant, which was verified using substrate analogs and inhibitors. Bioinformatic analysis indicates this NRPS-like glycine betaine reductase is highly conserved and widespread in fungi kingdom. Ge- netic knockout experiments confirmed its role in choline biosynthesis and maintaining glycine betaine homeostasis in fungi. Our findings demonstrate that the oxidative choline-glycine beta- ine degradation pathway can operate in a fully reversible fashion and provide new insights in understanding fungal choline metabolism. -

Presence of a Gene Encoding Choline Sulfatase in Sinorhizobium Meliloti Bet Operon: Choline-O-Sulfate Is Metabolized Into Glycine Betaine
Proc. Natl. Acad. Sci. USA Vol. 95, pp. 11394–11399, September 1998 Microbiology Presence of a gene encoding choline sulfatase in Sinorhizobium meliloti bet operon: Choline-O-sulfate is metabolized into glycine betaine MAGNE ØSTERÅS*, ERIC BONCOMPAGNI†,NADINE VINCENT,MARIE-CHRISTINE POGGI, AND DANIEL LE RUDULIER‡ Laboratoire de Biologie Ve´ge´tale et Microbiologie, Centre National de la Recherche Scientifique Equipe en Restructuration 590, Universite´de Nice–Sophia Antipolis, 06108 Nice Cedex, France Edited by Roland Douce, University of Grenoble, Grenoble, France, and approved July 17, 1998 (received for review April 24, 1998) ABSTRACT Glycine betaine is a potent osmoprotectant been shown that S. meliloti has the ability to use different accumulated by Sinorhizobium meliloti to cope with osmotic compatible solutes in presence of an osmotic stress, it appears that stress. The biosynthesis of glycine betaine from choline is en- glycine betaine is the most potent osmoprotectant and strongly coded by an operon of four genes, betICBA, as determined by stimulates the growth rate of the bacteria in high-salt medium (8). sequence and mutant analysis. The betI and betC genes are Unlike enteric bacteria such as Escherichia coli, S. meliloti also separated by an intergenic region containing a 130-bp mosaic catabolizes glycine betaine and uses it both as a carbon and a element that also is present between the betB and betA genes. In nitrogen source for growth (9, 10). The importance of this addition to the genes encoding a presumed regulatory protein compound in response to osmotic stress is confirmed by the (betI), the betaine aldehyde dehydrogenase (betB), and the cho- observation that the catabolic pathway is repressed in presence of line dehydrogenase (betA) enzymes also found in Escherichia coli, high-salt concentration, favoring the accumulation of the com- pound over its metabolization (10). -

Subtilis Temperature Stress Protection to Bacillus Dimethylglycine
Dimethylglycine Provides Salt and Temperature Stress Protection to Bacillus subtilis Abdallah Bashir, Tamara Hoffmann, Sander H. J. Smits and Downloaded from Erhard Bremer Appl. Environ. Microbiol. 2014, 80(9):2773. DOI: 10.1128/AEM.00078-14. Published Ahead of Print 21 February 2014. http://aem.asm.org/ Updated information and services can be found at: http://aem.asm.org/content/80/9/2773 These include: SUPPLEMENTAL MATERIAL Supplemental material on April 14, 2014 by UNIVERSITATSBIBLIOTHEK MARBURG REFERENCES This article cites 81 articles, 44 of which can be accessed free at: http://aem.asm.org/content/80/9/2773#ref-list-1 CONTENT ALERTS Receive: RSS Feeds, eTOCs, free email alerts (when new articles cite this article), more» Information about commercial reprint orders: http://journals.asm.org/site/misc/reprints.xhtml To subscribe to to another ASM Journal go to: http://journals.asm.org/site/subscriptions/ Dimethylglycine Provides Salt and Temperature Stress Protection to Bacillus subtilis Abdallah Bashir,a,b,c Tamara Hoffmann,a,d Sander H. J. Smits,e Erhard Bremera,d Downloaded from Laboratory for Microbiology, Department of Biology, Philipps-Universität Marburg, Marburg, Germanya; Al-Azhar University—Gaza, Faculty of Science, Biology Department, Gazab; Max Planck Institute for Terrestrial Microbiology, Emeritus Group of R. K. Thauer, Marburg, Germanyc; LOEWE Center for Synthetic Microbiology, Philipps-Universität Marburg, Marburg, Germanyd; Institute of Biochemistry, Heinrich Heine University Düsseldorf, Düsseldorf, Germanye Glycine betaine is a potent osmotic and thermal stress protectant of many microorganisms. Its synthesis from glycine results in the formation of the intermediates monomethylglycine (sarcosine) and dimethylglycine (DMG), and these compounds are also produced when it is catabolized. -

Mining the Red Sea Metagenomics Libraries for Betaine Pathways
School of Sciences and Engineering Mining the Red Sea Metagenomics Libraries for Betaine Pathways A Thesis Submitted to The Biotechnology Master's Program In partial fulfillment of the requirements for the degree of Master of Science By: Sherouk Abou Allam Under the supervision of: Dr. Walid Fouad 2018 I The American University in Cairo Mining the Red Sea Metagenomics Libraries for Betaine Pathways A Thesis Submitted by Sherouk Abou Allam To the Biotechnology Graduate Program 2018 In partial fulfillment of the requirements for The degree of Master of Science Has been approved by Thesis Committee Supervisor/Chair _______________________________________________ Affiliation ______________________________________ Thesis Committee Reader/Examiner ______________________________________________ Affiliation _____________________________________ Thesis Committee Reader/Examiner ______________________________________________ Affiliation ______________________________________ Thesis Committee Reader/External Examiner _______________________________________ Affiliation_______________________________________ ________________ _____________ _________________ _______________ Dept. Chair/Director Date Dean Date II DEDICATION To my beloved parents who taught me to love science and never lose hope. To Mokhtar, my husband, who has been a constant source of support and encouragement during the challenges of graduation. To Arwa and Salman, my children, who brought smiles to my life and gave me the reason to live. To Nissma, my sister, the best friend and best guidance who taught me faith. III ACKNOWLEDGEMENTS This work could have never been done without the help, support and guidance of Dr. Walid. I sincerely thank him for everything. Special thanks to Dr. Rania Siam, without the supportive words of her, I would not have continued my studies. Special thanks to Dr. Ahmed Moustafa, who taught me lots techniques of bioinformatics. I am very grateful to Mustafa Adel and Nahla Hussein for their help through the years of my graduate study. -
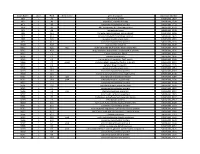
NTML 384-Well Array Well Identification
Strain Name plate Well Gene name gene discription Accession Number NE1 1 A1 cell surface protein SAUSA300_1327 NE2 1 A3 peptidase, rhomboid family SAUSA300_1509 NE3 1 A5 conserved hypothetical protein SAUSA300_1733 NE4 1 A7 ABC transporter ATP-binding protein SAUSA300_0309 NE5 1 A9 aminotransferase SAUSA300_2539 NE6 1 A11 formate dehydrogenase, alpha subunit SAUSA300_2258 NE7 1 A13 conserved hypothetical phage protein SAUSA300_1425 NE8 1 A15 putative membrane protein SAUSA300_1481 NE9 1 A17 conserved hypothetical protein SAUSA300_1294 NE10 1 A19 putative hemolysin III SAUSA300_2129 NE11 1 A21 recJ single-stranded-DNA-specific exonuclease RecJ SAUSA300_1592 NE12 1 A23 drug resistance transporter, EmrB/QacA subfamily SAUSA300_2126 NE13 1 C1 ribose transporter RbsU SAUSA300_0264 NE14 1 C3 putative transporter SAUSA300_2406 NE15 1 C5 transcriptional regulator, TetR family SAUSA300_2509 NE16 1 C7 moaD molybdopterin converting factor, subunit 1 SAUSA300_2221 NE17 1 C9 putative drug transporter SAUSA300_1705 NE18 1 C11 putative membrane protein SAUSA300_1851 NE19 1 C13 conserved hypothetical protein SAUSA300_0369 NE20 1 C15 transcriptional antiterminator, BglG family SAUSA300_0238 NE21 1 C17 upp uracil phosphoribosyltransferase SAUSA300_2066 NE22 1 C19 polA DNA polymerase I superfamily SAUSA300_1636 NE23 1 C21 conserved hypothetical protein SAUSA300_0740 NE24 1 C23 secretory antigen precursor SsaA SAUSA300_2503 NE25 1 E1 conserved hypothetical protein SAUSA300_1905 NE26 1 E3 coa staphylocoagulase precursor SAUSA300_0224 NE27 1 E5 nitric oxide -

Www .Alfa.Com
Bio 2013-14 Alfa Aesar North America Alfa Aesar Korea Uni-Onward (International Sales Headquarters) 101-3701, Lotte Castle President 3F-2 93 Wenhau 1st Rd, Sec 1, 26 Parkridge Road O-Dong Linkou Shiang 244, Taipei County Ward Hill, MA 01835 USA 467, Gongduk-Dong, Mapo-Gu Taiwan Tel: 1-800-343-0660 or 1-978-521-6300 Seoul, 121-805, Korea Tel: 886-2-2600-0611 Fax: 1-978-521-6350 Tel: +82-2-3140-6000 Fax: 886-2-2600-0654 Email: [email protected] Fax: +82-2-3140-6002 Email: [email protected] Email: [email protected] Alfa Aesar United Kingdom Echo Chemical Co. Ltd Shore Road Alfa Aesar India 16, Gongyeh Rd, Lu-Chu Li Port of Heysham Industrial Park (Johnson Matthey Chemicals India Toufen, 351, Miaoli Heysham LA3 2XY Pvt. Ltd.) Taiwan England Kandlakoya Village Tel: 866-37-629988 Bio Chemicals for Life Tel: 0800-801812 or +44 (0)1524 850506 Medchal Mandal Email: [email protected] www.alfa.com Fax: +44 (0)1524 850608 R R District Email: [email protected] Hyderabad - 501401 Andhra Pradesh, India Including: Alfa Aesar Germany Tel: +91 40 6730 1234 Postbox 11 07 65 Fax: +91 40 6730 1230 Amino Acids and Derivatives 76057 Karlsruhe Email: [email protected] Buffers Germany Tel: 800 4566 4566 or Distributed By: Click Chemistry Reagents +49 (0)721 84007 280 Electrophoresis Reagents Fax: +49 (0)721 84007 300 Hydrus Chemical Inc. Email: [email protected] Uchikanda 3-Chome, Chiyoda-Ku Signal Transduction Reagents Tokyo 101-0047 Western Blot and ELISA Reagents Alfa Aesar France Japan 2 allée d’Oslo Tel: 03(3258)5031 ...and much more 67300 Schiltigheim Fax: 03(3258)6535 France Email: [email protected] Tel: 0800 03 51 47 or +33 (0)3 8862 2690 Fax: 0800 10 20 67 or OOO “REAKOR” +33 (0)3 8862 6864 Nagorny Proezd, 7 Email: [email protected] 117 105 Moscow Russia Alfa Aesar China Tel: +7 495 640 3427 Room 1509 Fax: +7 495 640 3427 ext 6 CBD International Building Email: [email protected] No.