Proteomic and Transcriptomic Profiling Reveal Different Aspects of Aging In
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Screening and Identification of Key Biomarkers in Clear Cell Renal Cell Carcinoma Based on Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Screening and identification of key biomarkers in clear cell renal cell carcinoma based on bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Clear cell renal cell carcinoma (ccRCC) is one of the most common types of malignancy of the urinary system. The pathogenesis and effective diagnosis of ccRCC have become popular topics for research in the previous decade. In the current study, an integrated bioinformatics analysis was performed to identify core genes associated in ccRCC. An expression dataset (GSE105261) was downloaded from the Gene Expression Omnibus database, and included 26 ccRCC and 9 normal kideny samples. Assessment of the microarray dataset led to the recognition of differentially expressed genes (DEGs), which was subsequently used for pathway and gene ontology (GO) enrichment analysis. -

Small Cell Ovarian Carcinoma: Genomic Stability and Responsiveness to Therapeutics
Gamwell et al. Orphanet Journal of Rare Diseases 2013, 8:33 http://www.ojrd.com/content/8/1/33 RESEARCH Open Access Small cell ovarian carcinoma: genomic stability and responsiveness to therapeutics Lisa F Gamwell1,2, Karen Gambaro3, Maria Merziotis2, Colleen Crane2, Suzanna L Arcand4, Valerie Bourada1,2, Christopher Davis2, Jeremy A Squire6, David G Huntsman7,8, Patricia N Tonin3,4,5 and Barbara C Vanderhyden1,2* Abstract Background: The biology of small cell ovarian carcinoma of the hypercalcemic type (SCCOHT), which is a rare and aggressive form of ovarian cancer, is poorly understood. Tumourigenicity, in vitro growth characteristics, genetic and genomic anomalies, and sensitivity to standard and novel chemotherapeutic treatments were investigated in the unique SCCOHT cell line, BIN-67, to provide further insight in the biology of this rare type of ovarian cancer. Method: The tumourigenic potential of BIN-67 cells was determined and the tumours formed in a xenograft model was compared to human SCCOHT. DNA sequencing, spectral karyotyping and high density SNP array analysis was performed. The sensitivity of the BIN-67 cells to standard chemotherapeutic agents and to vesicular stomatitis virus (VSV) and the JX-594 vaccinia virus was tested. Results: BIN-67 cells were capable of forming spheroids in hanging drop cultures. When xenografted into immunodeficient mice, BIN-67 cells developed into tumours that reflected the hypercalcemia and histology of human SCCOHT, notably intense expression of WT-1 and vimentin, and lack of expression of inhibin. Somatic mutations in TP53 and the most common activating mutations in KRAS and BRAF were not found in BIN-67 cells by DNA sequencing. -

Viewed Under 23 (B) Or 203 (C) fi M M Male Cko Mice, and Largely Unaffected Magni Cation; Scale Bars, 500 M (B) and 50 M (C)
BRIEF COMMUNICATION www.jasn.org Renal Fanconi Syndrome and Hypophosphatemic Rickets in the Absence of Xenotropic and Polytropic Retroviral Receptor in the Nephron Camille Ansermet,* Matthias B. Moor,* Gabriel Centeno,* Muriel Auberson,* † † ‡ Dorothy Zhang Hu, Roland Baron, Svetlana Nikolaeva,* Barbara Haenzi,* | Natalya Katanaeva,* Ivan Gautschi,* Vladimir Katanaev,*§ Samuel Rotman, Robert Koesters,¶ †† Laurent Schild,* Sylvain Pradervand,** Olivier Bonny,* and Dmitri Firsov* BRIEF COMMUNICATION *Department of Pharmacology and Toxicology and **Genomic Technologies Facility, University of Lausanne, Lausanne, Switzerland; †Department of Oral Medicine, Infection, and Immunity, Harvard School of Dental Medicine, Boston, Massachusetts; ‡Institute of Evolutionary Physiology and Biochemistry, St. Petersburg, Russia; §School of Biomedicine, Far Eastern Federal University, Vladivostok, Russia; |Services of Pathology and ††Nephrology, Department of Medicine, University Hospital of Lausanne, Lausanne, Switzerland; and ¶Université Pierre et Marie Curie, Paris, France ABSTRACT Tight control of extracellular and intracellular inorganic phosphate (Pi) levels is crit- leaves.4 Most recently, Legati et al. have ical to most biochemical and physiologic processes. Urinary Pi is freely filtered at the shown an association between genetic kidney glomerulus and is reabsorbed in the renal tubule by the action of the apical polymorphisms in Xpr1 and primary fa- sodium-dependent phosphate transporters, NaPi-IIa/NaPi-IIc/Pit2. However, the milial brain calcification disorder.5 How- molecular identity of the protein(s) participating in the basolateral Pi efflux remains ever, the role of XPR1 in the maintenance unknown. Evidence has suggested that xenotropic and polytropic retroviral recep- of Pi homeostasis remains unknown. Here, tor 1 (XPR1) might be involved in this process. Here, we show that conditional in- we addressed this issue in mice deficient for activation of Xpr1 in the renal tubule in mice resulted in impaired renal Pi Xpr1 in the nephron. -

(SLC22A18) (NM 183233) Human Recombinant Protein Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TP304889 Solute carrier family 22 member 18 (SLC22A18) (NM_183233) Human Recombinant Protein Product data: Product Type: Recombinant Proteins Description: Recombinant protein of human solute carrier family 22, member 18 (SLC22A18), transcript variant 2 Species: Human Expression Host: HEK293T Tag: C-Myc/DDK Predicted MW: 44.7 kDa Concentration: >50 ug/mL as determined by microplate BCA method Purity: > 80% as determined by SDS-PAGE and Coomassie blue staining Buffer: 25 mM Tris.HCl, pH 7.3, 100 mM glycine, 10% glycerol Preparation: Recombinant protein was captured through anti-DDK affinity column followed by conventional chromatography steps. Storage: Store at -80°C. Stability: Stable for 12 months from the date of receipt of the product under proper storage and handling conditions. Avoid repeated freeze-thaw cycles. RefSeq: NP_899056 Locus ID: 5002 UniProt ID: Q96BI1 RefSeq Size: 1563 Cytogenetics: 11p15.4 RefSeq ORF: 1272 Synonyms: BWR1A; BWSCR1A; HET; IMPT1; ITM; ORCTL2; p45-BWR1A; SLC22A1L; TSSC5 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 Solute carrier family 22 member 18 (SLC22A18) (NM_183233) Human Recombinant Protein – TP304889 Summary: This gene is one of several tumor-suppressing subtransferable fragments located in the imprinted gene domain of 11p15.5, an important tumor-suppressor gene region. Alterations in this region have been associated with the Beckwith-Wiedemann syndrome, Wilms tumor, rhabdomyosarcoma, adrenocortical carcinoma, and lung, ovarian, and breast cancer. -

The Role of the Mtor Pathway in Developmental Reprogramming Of
THE ROLE OF THE MTOR PATHWAY IN DEVELOPMENTAL REPROGRAMMING OF HEPATIC LIPID METABOLISM AND THE HEPATIC TRANSCRIPTOME AFTER EXPOSURE TO 2,2',4,4'- TETRABROMODIPHENYL ETHER (BDE-47) An Honors Thesis Presented By JOSEPH PAUL MCGAUNN Approved as to style and content by: ________________________________________________________** Alexander Suvorov 05/18/20 10:40 ** Chair ________________________________________________________** Laura V Danai 05/18/20 10:51 ** Committee Member ________________________________________________________** Scott C Garman 05/18/20 10:57 ** Honors Program Director ABSTRACT An emerging hypothesis links the epidemic of metabolic diseases, such as non-alcoholic fatty liver disease (NAFLD) and diabetes with chemical exposures during development. Evidence from our lab and others suggests that developmental exposure to environmentally prevalent flame-retardant BDE47 may permanently reprogram hepatic lipid metabolism, resulting in an NAFLD-like phenotype. Additionally, we have demonstrated that BDE-47 alters the activity of both mTOR complexes (mTORC1 and 2) in hepatocytes. The mTOR pathway integrates environmental information from different signaling pathways, and regulates key cellular functions such as lipid metabolism, innate immunity, and ribosome biogenesis. Thus, we hypothesized that the developmental effects of BDE-47 on liver lipid metabolism are mTOR-dependent. To assess this, we generated mice with liver-specific deletions of mTORC1 or mTORC2 and exposed these mice and their respective controls perinatally to -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -
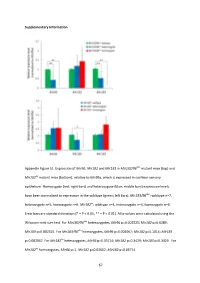
And Mir183 in Mir183/96 Dko Mutant Mice (Top) And
Supplementary Information Appendix Figure S1. Expression of Mir96 , Mir182 and Mir183 in Mir183/96 dko mutant mice (top) and Mir182 ko mutant mice (bottom), relative to Mir99a , which is expressed in cochlear sensory epithelium. Homozygote (red; right bars) and heterozygote (blue; middle bars) expression levels have been normalised to expression in the wildtype (green; left bars). Mir183/96 dko : wildtype n=7, heterozygote n=5, homozygote n=6. Mir182 ko : wildtype n=4, heterozygote n=4, homozygote n=4. Error bars are standard deviation (* = P < 0.05, ** = P < 0.01). All p-values were calculated using the Wilcoxon rank sum test. For Mir183/96 dko heterozygotes, Mir96 p=0.002525; Mir182 p=0.6389; Mir183 p=0.002525. For Mir183/96 dko homozygotes, Mir96 p=0.002067; Mir182 p=0.1014; Mir183 p=0.002067. For Mir182 ko heterozygotes, Mir96 p=0.05714; Mir182 p=0.3429; Mir183 p=0.3429. For Mir182 ko homozygotes, Mir96 p=1; Mir182 p=0.02652; Mir183 p=0.05714. 67 68 Appendix Figure S2. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir183/96 dko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 69 70 Appendix Figure S3. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir182 ko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 71 Appendix Figure S4. Mean ABR waveforms at 12kHz, shown at 20dB (top) and 50dB (bottom) above threshold (sensation level, SL) ± standard deviation, at four weeks old. -

Prestinłs Anion Transport and Voltage-Sensing Capabilities Are
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Biophysical Journal Volume 96 April 2009 3179–3186 3179 Prestin’s Anion Transport and Voltage-Sensing Capabilities Are Independent Jun-Ping Bai,† Alexei Surguchev,†‡ Simone Montoya,†‡ Peter S. Aronson,§{ Joseph Santos-Sacchi,‡{k and Dhasakumar Navaratnam†k* †Department of Neurology, ‡Division of Otolaryngology, Department of Surgery, §Department of Internal Medicine, {Department of Cellular and Molecular Physiology, and kDepartment of Neurobiology, Yale University School of Medicine, New Haven, Connecticut ABSTRACT The integral membrane protein prestin, a member of the SLC26 anion transporter family, is responsible for the voltage-driven electromotility of mammalian outer hair cells. It was argued that the evolution of prestin’s motor function required a loss of the protein’s transport capabilities. Instead, it was proposed that prestin manages only an abortive hemicycle that results in the trapped anion acting as a voltage sensor, to generate the motor’s signature gating charge movement or nonlinear capaci- tance. We demonstrate, using classical radioactive anion ([14C]formate and [14C]oxalate) uptake studies, that in contrast to previous observations, prestin is able to transport anions. The prestin-dependent uptake of both these anions was twofold that of cells transfected with vector alone, and comparable to SLC26a6, prestin’s closest phylogenetic relative. Furthermore, we identify a potential chloride-binding site in which the mutations of two residues (P328A and L326A) preserve nonlinear capacitance, yet negate anion transport. Finally, we distinguish 12 charged residues out of 22, residing within prestin’s transmembrane regions, that contribute to unitary charge movement, i.e., voltage sensing. -

Anti-Inflammatory Role of Curcumin in LPS Treated A549 Cells at Global Proteome Level and on Mycobacterial Infection
Anti-inflammatory Role of Curcumin in LPS Treated A549 cells at Global Proteome level and on Mycobacterial infection. Suchita Singh1,+, Rakesh Arya2,3,+, Rhishikesh R Bargaje1, Mrinal Kumar Das2,4, Subia Akram2, Hossain Md. Faruquee2,5, Rajendra Kumar Behera3, Ranjan Kumar Nanda2,*, Anurag Agrawal1 1Center of Excellence for Translational Research in Asthma and Lung Disease, CSIR- Institute of Genomics and Integrative Biology, New Delhi, 110025, India. 2Translational Health Group, International Centre for Genetic Engineering and Biotechnology, New Delhi, 110067, India. 3School of Life Sciences, Sambalpur University, Jyoti Vihar, Sambalpur, Orissa, 768019, India. 4Department of Respiratory Sciences, #211, Maurice Shock Building, University of Leicester, LE1 9HN 5Department of Biotechnology and Genetic Engineering, Islamic University, Kushtia- 7003, Bangladesh. +Contributed equally for this work. S-1 70 G1 S 60 G2/M 50 40 30 % of cells 20 10 0 CURI LPSI LPSCUR Figure S1: Effect of curcumin and/or LPS treatment on A549 cell viability A549 cells were treated with curcumin (10 µM) and/or LPS or 1 µg/ml for the indicated times and after fixation were stained with propidium iodide and Annexin V-FITC. The DNA contents were determined by flow cytometry to calculate percentage of cells present in each phase of the cell cycle (G1, S and G2/M) using Flowing analysis software. S-2 Figure S2: Total proteins identified in all the three experiments and their distribution betwee curcumin and/or LPS treated conditions. The proteins showing differential expressions (log2 fold change≥2) in these experiments were presented in the venn diagram and certain number of proteins are common in all three experiments. -

KCC4/SLC12A7 Polyclonal Antibody Catalog Number:26590-1-AP
For Research Use Only KCC4/SLC12A7 Polyclonal antibody www.ptglab.com Catalog Number:26590-1-AP Catalog Number: GenBank Accession Number: Purification Method: Basic Information 26590-1-AP BC098390 Antigen affinity purification Size: GeneID (NCBI): Recommended Dilutions: 150ul , Concentration: 900 μg/ml by 10723 WB 1:2000-1:10000 Nanodrop; Full Name: Source: solute carrier family 12 Rabbit (potassium/chloride transporters), Isotype: member 7 IgG Immunogen Catalog Number: AG24954 Applications Tested Applications: Positive Controls: WB, ELISA WB : BGC-823 cells, NCI-H1299 cells, Raji cells, SH- Species Specificity: SY5Y cells human KCC4, also named as Solute carrier family 12 member 7(SLC12A7), is a 1083 amino-acid protein, which belongs to Background Information the SLC12A transporter family. As a multi-pass membrane protein, the molecular weight of KCC4 is 119 kDa. KCC4 mediates electroneutral potassium-chloride cotransport when activated by cell swelling. Moreover, KCC4 may mediate K+ uptake into Deiters' cells in the cochlea and contribute to K+ recycling in the inner ear. KCC4 is important for the survival of cochlear outer and inner hair cells and the maintenance of the organ of Corti(Uniprot, GeneID:10723). Storage: Storage Store at -20°C. Stable for one year after shipment. Storage Buffer: PBS with 0.02% sodium azide and 50% glycerol pH 7.3. Aliquoting is unnecessary for -20ºC storage For technical support and original validation data for this product please contact: This product is exclusively available under Proteintech T: 1 (888) 4PTGLAB (1-888-478-4522) (toll free E: [email protected] Group brand and is not available to purchase from any in USA), or 1(312) 455-8498 (outside USA) W: ptglab.com other manufacturer. -

Identification of Transcriptomic Differences Between Lower
International Journal of Molecular Sciences Article Identification of Transcriptomic Differences between Lower Extremities Arterial Disease, Abdominal Aortic Aneurysm and Chronic Venous Disease in Peripheral Blood Mononuclear Cells Specimens Daniel P. Zalewski 1,*,† , Karol P. Ruszel 2,†, Andrzej St˛epniewski 3, Dariusz Gałkowski 4, Jacek Bogucki 5 , Przemysław Kołodziej 6 , Jolanta Szyma ´nska 7 , Bartosz J. Płachno 8 , Tomasz Zubilewicz 9 , Marcin Feldo 9,‡ , Janusz Kocki 2,‡ and Anna Bogucka-Kocka 1,‡ 1 Chair and Department of Biology and Genetics, Medical University of Lublin, 4a Chod´zkiSt., 20-093 Lublin, Poland; [email protected] 2 Chair of Medical Genetics, Department of Clinical Genetics, Medical University of Lublin, 11 Radziwiłłowska St., 20-080 Lublin, Poland; [email protected] (K.P.R.); [email protected] (J.K.) 3 Ecotech Complex Analytical and Programme Centre for Advanced Environmentally Friendly Technologies, University of Marie Curie-Skłodowska, 39 Gł˛ebokaSt., 20-612 Lublin, Poland; [email protected] 4 Department of Pathology and Laboratory Medicine, Rutgers-Robert Wood Johnson Medical School, One Robert Wood Johnson Place, New Brunswick, NJ 08903-0019, USA; [email protected] 5 Chair and Department of Organic Chemistry, Medical University of Lublin, 4a Chod´zkiSt., Citation: Zalewski, D.P.; Ruszel, K.P.; 20-093 Lublin, Poland; [email protected] St˛epniewski,A.; Gałkowski, D.; 6 Laboratory of Diagnostic Parasitology, Chair and Department of Biology and Genetics, Medical University of Bogucki, J.; Kołodziej, P.; Szyma´nska, Lublin, 4a Chod´zkiSt., 20-093 Lublin, Poland; [email protected] J.; Płachno, B.J.; Zubilewicz, T.; Feldo, 7 Department of Integrated Paediatric Dentistry, Chair of Integrated Dentistry, Medical University of Lublin, M.; et al. -

Supplementary Table S1. Correlation Between the Mutant P53-Interacting Partners and PTTG3P, PTTG1 and PTTG2, Based on Data from Starbase V3.0 Database
Supplementary Table S1. Correlation between the mutant p53-interacting partners and PTTG3P, PTTG1 and PTTG2, based on data from StarBase v3.0 database. PTTG3P PTTG1 PTTG2 Gene ID Coefficient-R p-value Coefficient-R p-value Coefficient-R p-value NF-YA ENSG00000001167 −0.077 8.59e-2 −0.210 2.09e-6 −0.122 6.23e-3 NF-YB ENSG00000120837 0.176 7.12e-5 0.227 2.82e-7 0.094 3.59e-2 NF-YC ENSG00000066136 0.124 5.45e-3 0.124 5.40e-3 0.051 2.51e-1 Sp1 ENSG00000185591 −0.014 7.50e-1 −0.201 5.82e-6 −0.072 1.07e-1 Ets-1 ENSG00000134954 −0.096 3.14e-2 −0.257 4.83e-9 0.034 4.46e-1 VDR ENSG00000111424 −0.091 4.10e-2 −0.216 1.03e-6 0.014 7.48e-1 SREBP-2 ENSG00000198911 −0.064 1.53e-1 −0.147 9.27e-4 −0.073 1.01e-1 TopBP1 ENSG00000163781 0.067 1.36e-1 0.051 2.57e-1 −0.020 6.57e-1 Pin1 ENSG00000127445 0.250 1.40e-8 0.571 9.56e-45 0.187 2.52e-5 MRE11 ENSG00000020922 0.063 1.56e-1 −0.007 8.81e-1 −0.024 5.93e-1 PML ENSG00000140464 0.072 1.05e-1 0.217 9.36e-7 0.166 1.85e-4 p63 ENSG00000073282 −0.120 7.04e-3 −0.283 1.08e-10 −0.198 7.71e-6 p73 ENSG00000078900 0.104 2.03e-2 0.258 4.67e-9 0.097 3.02e-2 Supplementary Table S2.