In-Memory Genomics Data Processing Through Apache Arrow Framework
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mastering Machine Learning with Scikit-Learn
www.it-ebooks.info Mastering Machine Learning with scikit-learn Apply effective learning algorithms to real-world problems using scikit-learn Gavin Hackeling BIRMINGHAM - MUMBAI www.it-ebooks.info Mastering Machine Learning with scikit-learn Copyright © 2014 Packt Publishing All rights reserved. No part of this book may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, without the prior written permission of the publisher, except in the case of brief quotations embedded in critical articles or reviews. Every effort has been made in the preparation of this book to ensure the accuracy of the information presented. However, the information contained in this book is sold without warranty, either express or implied. Neither the author, nor Packt Publishing, and its dealers and distributors will be held liable for any damages caused or alleged to be caused directly or indirectly by this book. Packt Publishing has endeavored to provide trademark information about all of the companies and products mentioned in this book by the appropriate use of capitals. However, Packt Publishing cannot guarantee the accuracy of this information. First published: October 2014 Production reference: 1221014 Published by Packt Publishing Ltd. Livery Place 35 Livery Street Birmingham B3 2PB, UK. ISBN 978-1-78398-836-5 www.packtpub.com Cover image by Amy-Lee Winfield [email protected]( ) www.it-ebooks.info Credits Author Project Coordinator Gavin Hackeling Danuta Jones Reviewers Proofreaders Fahad Arshad Simran Bhogal Sarah -

Installing Python Ø Basics of Programming • Data Types • Functions • List Ø Numpy Library Ø Pandas Library What Is Python?
Python in Data Science Programming Ha Khanh Nguyen Agenda Ø What is Python? Ø The Python Ecosystem • Installing Python Ø Basics of Programming • Data Types • Functions • List Ø NumPy Library Ø Pandas Library What is Python? • “Python is an interpreted high-level general-purpose programming language.” – Wikipedia • It supports multiple programming paradigms, including: • Structured/procedural • Object-oriented • Functional The Python Ecosystem Source: Fabien Maussion’s “Getting started with Python” Workshop Installing Python • Install Python through Anaconda/Miniconda. • This allows you to create and work in Python environments. • You can create multiple environments as needed. • Highly recommended: install Python through Miniconda. • Are we ready to “play” with Python yet? • Almost! • Most data scientists use Python through Jupyter Notebook, a web application that allows you to create a virtual notebook containing both text and code! • Python Installation tutorial: [Mac OS X] [Windows] For today’s seminar • Due to the time limitation, we will be using Google Colab instead. • Google Colab is a free Jupyter notebook environment that runs entirely in the cloud, so you can run Python code, write report in Jupyter Notebook even without installing the Python ecosystem. • It is NOT a complete alternative to installing Python on your local computer. • But it is a quick and easy way to get started/try out Python. • You will need to log in using your university account (possible for some schools) or your personal Google account. Basics of Programming Indentations – Not Braces • Other languages (R, C++, Java, etc.) use braces to structure code. # R code (not Python) a = 10 if (a < 5) { result = TRUE b = 0 } else { result = FALSE b = 100 } a = 10 if (a < 5) { result = TRUE; b = 0} else {result = FALSE; b = 100} Basics of Programming Indentations – Not Braces • Python uses whitespaces (tabs or spaces) to structure code instead. -

Quick Install for AWS EMR
Quick Install for AWS EMR Version: 6.8 Doc Build Date: 01/21/2020 Copyright © Trifacta Inc. 2020 - All Rights Reserved. CONFIDENTIAL These materials (the “Documentation”) are the confidential and proprietary information of Trifacta Inc. and may not be reproduced, modified, or distributed without the prior written permission of Trifacta Inc. EXCEPT AS OTHERWISE PROVIDED IN AN EXPRESS WRITTEN AGREEMENT, TRIFACTA INC. PROVIDES THIS DOCUMENTATION AS-IS AND WITHOUT WARRANTY AND TRIFACTA INC. DISCLAIMS ALL EXPRESS AND IMPLIED WARRANTIES TO THE EXTENT PERMITTED, INCLUDING WITHOUT LIMITATION THE IMPLIED WARRANTIES OF MERCHANTABILITY, NON-INFRINGEMENT AND FITNESS FOR A PARTICULAR PURPOSE AND UNDER NO CIRCUMSTANCES WILL TRIFACTA INC. BE LIABLE FOR ANY AMOUNT GREATER THAN ONE HUNDRED DOLLARS ($100) BASED ON ANY USE OF THE DOCUMENTATION. For third-party license information, please select About Trifacta from the Help menu. 1. Release Notes . 4 1.1 Changes to System Behavior . 4 1.1.1 Changes to the Language . 4 1.1.2 Changes to the APIs . 18 1.1.3 Changes to Configuration 23 1.1.4 Changes to the Object Model . 26 1.2 Release Notes 6.8 . 30 1.3 Release Notes 6.4 . 36 1.4 Release Notes 6.0 . 42 1.5 Release Notes 5.1 . 49 2. Quick Start 55 2.1 Install from AWS Marketplace with EMR . 55 2.2 Upgrade for AWS Marketplace with EMR . 62 3. Configure 62 3.1 Configure for AWS . 62 3.1.1 Configure for EC2 Role-Based Authentication . 68 3.1.2 Enable S3 Access . 70 3.1.2.1 Create Redshift Connections 81 3.1.3 Configure for EMR . -

Delft University of Technology Arrowsam In-Memory Genomics
Delft University of Technology ArrowSAM In-Memory Genomics Data Processing Using Apache Arrow Ahmad, Tanveer; Ahmed, Nauman; Peltenburg, Johan; Al-Ars, Zaid DOI 10.1109/ICCAIS48893.2020.9096725 Publication date 2020 Document Version Accepted author manuscript Published in 2020 3rd International Conference on Computer Applications & Information Security (ICCAIS) Citation (APA) Ahmad, T., Ahmed, N., Peltenburg, J., & Al-Ars, Z. (2020). ArrowSAM: In-Memory Genomics Data Processing Using Apache Arrow. In 2020 3rd International Conference on Computer Applications & Information Security (ICCAIS): Proceedings (pp. 1-6). [9096725] IEEE . https://doi.org/10.1109/ICCAIS48893.2020.9096725 Important note To cite this publication, please use the final published version (if applicable). Please check the document version above. Copyright Other than for strictly personal use, it is not permitted to download, forward or distribute the text or part of it, without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license such as Creative Commons. Takedown policy Please contact us and provide details if you believe this document breaches copyrights. We will remove access to the work immediately and investigate your claim. This work is downloaded from Delft University of Technology. For technical reasons the number of authors shown on this cover page is limited to a maximum of 10. © 2020 IEEE. Personal use of this material is permitted. Permission from IEEE must be obtained for all other uses, in any current or future media, including reprinting/republishing this material for advertising or promotional purposes, creating new collective works, for resale or redistribution to servers or lists, or reuse of any copyrighted component of this work in other works. -

The Platform Inside and out Release 0.8
The Platform Inside and Out Release 0.8 Joshua Patterson – GM, Data Science RAPIDS End-to-End Accelerated GPU Data Science Data Preparation Model Training Visualization Dask cuDF cuIO cuML cuGraph PyTorch Chainer MxNet cuXfilter <> pyViz Analytics Machine Learning Graph Analytics Deep Learning Visualization GPU Memory 2 Data Processing Evolution Faster data access, less data movement Hadoop Processing, Reading from disk HDFS HDFS HDFS HDFS HDFS Read Query Write Read ETL Write Read ML Train Spark In-Memory Processing 25-100x Improvement Less code HDFS Language flexible Read Query ETL ML Train Primarily In-Memory Traditional GPU Processing 5-10x Improvement More code HDFS GPU CPU GPU CPU GPU ML Language rigid Query ETL Read Read Write Read Write Read Train Substantially on GPU 3 Data Movement and Transformation The bane of productivity and performance APP B Read Data APP B GPU APP B Copy & Convert Data CPU GPU Copy & Convert Copy & Convert APP A GPU Data APP A Load Data APP A 4 Data Movement and Transformation What if we could keep data on the GPU? APP B Read Data APP B GPU APP B Copy & Convert Data CPU GPU Copy & Convert Copy & Convert APP A GPU Data APP A Load Data APP A 5 Learning from Apache Arrow ● Each system has its own internal memory format ● All systems utilize the same memory format ● 70-80% computation wasted on serialization and deserialization ● No overhead for cross-system communication ● Similar functionality implemented in multiple projects ● Projects can share functionality (eg, Parquet-to-Arrow reader) From Apache Arrow -
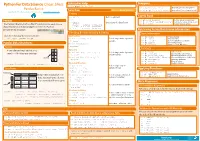
Cheat Sheet Pandas Python.Indd
Python For Data Science Cheat Sheet Asking For Help Dropping >>> help(pd.Series.loc) Pandas Basics >>> s.drop(['a', 'c']) Drop values from rows (axis=0) Selection Also see NumPy Arrays >>> df.drop('Country', axis=1) Drop values from columns(axis=1) Learn Python for Data Science Interactively at www.DataCamp.com Getting Sort & Rank >>> s['b'] Get one element -5 Pandas >>> df.sort_index() Sort by labels along an axis Get subset of a DataFrame >>> df.sort_values(by='Country') Sort by the values along an axis The Pandas library is built on NumPy and provides easy-to-use >>> df[1:] Assign ranks to entries Country Capital Population >>> df.rank() data structures and data analysis tools for the Python 1 India New Delhi 1303171035 programming language. 2 Brazil Brasília 207847528 Retrieving Series/DataFrame Information Selecting, Boolean Indexing & Setting Basic Information Use the following import convention: By Position (rows,columns) Select single value by row & >>> df.shape >>> import pandas as pd >>> df.iloc[[0],[0]] >>> df.index Describe index Describe DataFrame columns 'Belgium' column >>> df.columns Pandas Data Structures >>> df.info() Info on DataFrame >>> df.iat([0],[0]) Number of non-NA values >>> df.count() Series 'Belgium' Summary A one-dimensional labeled array a 3 By Label Select single value by row & >>> df.sum() Sum of values capable of holding any data type b -5 >>> df.loc[[0], ['Country']] Cummulative sum of values 'Belgium' column labels >>> df.cumsum() >>> df.min()/df.max() Minimum/maximum values c 7 Minimum/Maximum index -

Pandas: Powerful Python Data Analysis Toolkit Release 0.25.3
pandas: powerful Python data analysis toolkit Release 0.25.3 Wes McKinney& PyData Development Team Nov 02, 2019 CONTENTS i ii pandas: powerful Python data analysis toolkit, Release 0.25.3 Date: Nov 02, 2019 Version: 0.25.3 Download documentation: PDF Version | Zipped HTML Useful links: Binary Installers | Source Repository | Issues & Ideas | Q&A Support | Mailing List pandas is an open source, BSD-licensed library providing high-performance, easy-to-use data structures and data analysis tools for the Python programming language. See the overview for more detail about whats in the library. CONTENTS 1 pandas: powerful Python data analysis toolkit, Release 0.25.3 2 CONTENTS CHAPTER ONE WHATS NEW IN 0.25.2 (OCTOBER 15, 2019) These are the changes in pandas 0.25.2. See release for a full changelog including other versions of pandas. Note: Pandas 0.25.2 adds compatibility for Python 3.8 (GH28147). 1.1 Bug fixes 1.1.1 Indexing • Fix regression in DataFrame.reindex() not following the limit argument (GH28631). • Fix regression in RangeIndex.get_indexer() for decreasing RangeIndex where target values may be improperly identified as missing/present (GH28678) 1.1.2 I/O • Fix regression in notebook display where <th> tags were missing for DataFrame.index values (GH28204). • Regression in to_csv() where writing a Series or DataFrame indexed by an IntervalIndex would incorrectly raise a TypeError (GH28210) • Fix to_csv() with ExtensionArray with list-like values (GH28840). 1.1.3 Groupby/resample/rolling • Bug incorrectly raising an IndexError when passing a list of quantiles to pandas.core.groupby. DataFrameGroupBy.quantile() (GH28113). -

Arrow: Integration to 'Apache' 'Arrow'
Package ‘arrow’ September 5, 2021 Title Integration to 'Apache' 'Arrow' Version 5.0.0.2 Description 'Apache' 'Arrow' <https://arrow.apache.org/> is a cross-language development platform for in-memory data. It specifies a standardized language-independent columnar memory format for flat and hierarchical data, organized for efficient analytic operations on modern hardware. This package provides an interface to the 'Arrow C++' library. Depends R (>= 3.3) License Apache License (>= 2.0) URL https://github.com/apache/arrow/, https://arrow.apache.org/docs/r/ BugReports https://issues.apache.org/jira/projects/ARROW/issues Encoding UTF-8 Language en-US SystemRequirements C++11; for AWS S3 support on Linux, libcurl and openssl (optional) Biarch true Imports assertthat, bit64 (>= 0.9-7), methods, purrr, R6, rlang, stats, tidyselect, utils, vctrs RoxygenNote 7.1.1.9001 VignetteBuilder knitr Suggests decor, distro, dplyr, hms, knitr, lubridate, pkgload, reticulate, rmarkdown, stringi, stringr, testthat, tibble, withr Collate 'arrowExports.R' 'enums.R' 'arrow-package.R' 'type.R' 'array-data.R' 'arrow-datum.R' 'array.R' 'arrow-tabular.R' 'buffer.R' 'chunked-array.R' 'io.R' 'compression.R' 'scalar.R' 'compute.R' 'config.R' 'csv.R' 'dataset.R' 'dataset-factory.R' 'dataset-format.R' 'dataset-partition.R' 'dataset-scan.R' 'dataset-write.R' 'deprecated.R' 'dictionary.R' 'dplyr-arrange.R' 'dplyr-collect.R' 'dplyr-eval.R' 'dplyr-filter.R' 'expression.R' 'dplyr-functions.R' 1 2 R topics documented: 'dplyr-group-by.R' 'dplyr-mutate.R' 'dplyr-select.R' 'dplyr-summarize.R' -

Image Processing with Scikit-Image Emmanuelle Gouillart
Image processing with scikit-image Emmanuelle Gouillart Surface, Glass and Interfaces, CNRS/Saint-Gobain Paris-Saclay Center for Data Science Image processing Manipulating images in order to retrieve new images or image characteristics (features, measurements, ...) Often combined with machine learning Principle Some features The world is getting more and more visual Image processing Manipulating images in order to retrieve new images or image characteristics (features, measurements, ...) Often combined with machine learning Principle Some features The world is getting more and more visual Image processing Manipulating images in order to retrieve new images or image characteristics (features, measurements, ...) Often combined with machine learning Principle Some features The world is getting more and more visual Image processing Manipulating images in order to retrieve new images or image characteristics (features, measurements, ...) Often combined with machine learning Principle Some features The world is getting more and more visual Image processing Manipulating images in order to retrieve new images or image characteristics (features, measurements, ...) Often combined with machine learning Principle Some features The world is getting more and more visual Principle Some features The world is getting more and more visual Image processing Manipulating images in order to retrieve new images or image characteristics (features, measurements, ...) Often combined with machine learning Principle Some features scikit-image http://scikit-image.org/ A module of the Scientific Python stack Language: Python Core modules: NumPy, SciPy, matplotlib Application modules: scikit-learn, scikit-image, pandas, ... A general-purpose image processing library open-source (BSD) not an application (ImageJ) less specialized than other libraries (e.g. OpenCV for computer vision) 1 Principle Principle Some features 1 First steps from skimage import data , io , filter image = data . -

In Reference to RPC: It's Time to Add Distributed Memory
In Reference to RPC: It’s Time to Add Distributed Memory Stephanie Wang, Benjamin Hindman, Ion Stoica UC Berkeley ACM Reference Format: D (e.g., Apache Spark) F (e.g., Distributed TF) Stephanie Wang, Benjamin Hindman, Ion Stoica. 2021. In Refer- A B C i ii 1 2 3 ence to RPC: It’s Time to Add Distributed Memory. In Workshop RPC E on Hot Topics in Operating Systems (HotOS ’21), May 31-June 2, application 2021, Ann Arbor, MI, USA. ACM, New York, NY, USA, 8 pages. Figure 1: A single “application” actually consists of many https://doi.org/10.1145/3458336.3465302 components and distinct frameworks. With no shared address space, data (squares) must be copied between 1 Introduction different components. RPC has been remarkably successful. Most distributed ap- Load balancer Scheduler + Mem Mgt plications built today use an RPC runtime such as gRPC [3] Executors or Apache Thrift [2]. The key behind RPC’s success is the Servers request simple but powerful semantics of its programming model. client data request cache In particular, RPC has no shared state: arguments and return Distributed object store values are passed by value between processes, meaning that (a) (b) they must be copied into the request or reply. Thus, argu- ments and return values are inherently immutable. These Figure 2: Logical RPC architecture: (a) today, and (b) with a shared address space and automatic memory management. simple semantics facilitate highly efficient and reliable imple- mentations, as no distributed coordination is required, while then return a reference, i.e., some metadata that acts as a remaining useful for a general set of distributed applications. -

Ray: a Distributed Framework for Emerging AI Applications
Ray: A Distributed Framework for Emerging AI Applications Philipp Moritz, Robert Nishihara, Stephanie Wang, Alexey Tumanov, Richard Liaw, Eric Liang, Melih Elibol, Zongheng Yang, William Paul, Michael I. Jordan, and Ion Stoica, UC Berkeley https://www.usenix.org/conference/osdi18/presentation/nishihara This paper is included in the Proceedings of the 13th USENIX Symposium on Operating Systems Design and Implementation (OSDI ’18). October 8–10, 2018 • Carlsbad, CA, USA ISBN 978-1-939133-08-3 Open access to the Proceedings of the 13th USENIX Symposium on Operating Systems Design and Implementation is sponsored by USENIX. Ray: A Distributed Framework for Emerging AI Applications Philipp Moritz,∗ Robert Nishihara,∗ Stephanie Wang, Alexey Tumanov, Richard Liaw, Eric Liang, Melih Elibol, Zongheng Yang, William Paul, Michael I. Jordan, Ion Stoica University of California, Berkeley Abstract and their use in prediction. These frameworks often lever- age specialized hardware (e.g., GPUs and TPUs), with the The next generation of AI applications will continuously goal of reducing training time in a batch setting. Examples interact with the environment and learn from these inter- include TensorFlow [7], MXNet [18], and PyTorch [46]. actions. These applications impose new and demanding The promise of AI is, however, far broader than classi- systems requirements, both in terms of performance and cal supervised learning. Emerging AI applications must flexibility. In this paper, we consider these requirements increasingly operate in dynamic environments, react to and present Ray—a distributed system to address them. changes in the environment, and take sequences of ac- Ray implements a unified interface that can express both tions to accomplish long-term goals [8, 43]. -

Zero-Cost, Arrow-Enabled Data Interface for Apache Spark
Zero-Cost, Arrow-Enabled Data Interface for Apache Spark Sebastiaan Jayjeet Aaron Chu Ivo Jimenez Jeff LeFevre Alvarez Chakraborty UC Santa Cruz UC Santa Cruz UC Santa Cruz Rodriguez UC Santa Cruz [email protected] [email protected] [email protected] Leiden University [email protected] [email protected] Carlos Alexandru Uta Maltzahn Leiden University UC Santa Cruz [email protected] [email protected] ABSTRACT which is a very expensive operation; (2) data processing sys- Distributed data processing ecosystems are widespread and tems require new adapters or readers for each new data type their components are highly specialized, such that efficient to support and for each new system to integrate with. interoperability is urgent. Recently, Apache Arrow was cho- A common example where these two issues occur is the sen by the community to serve as a format mediator, provid- de-facto standard data processing engine, Apache Spark. In ing efficient in-memory data representation. Arrow enables Spark, the common data representation passed between op- efficient data movement between data processing and storage erators is row-based [5]. Connecting Spark to other systems engines, significantly improving interoperability and overall such as MongoDB [8], Azure SQL [7], Snowflake [11], or performance. In this work, we design a new zero-cost data in- data sources such as Parquet [30] or ORC [3], entails build- teroperability layer between Apache Spark and Arrow-based ing connectors and converting data. Although Spark was data sources through the Arrow Dataset API. Our novel data initially designed as a computation engine, this data adapter interface helps separate the computation (Spark) and data ecosystem was necessary to enable new types of workloads.