Delft University of Technology Arrowsam In-Memory Genomics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Quick Install for AWS EMR
Quick Install for AWS EMR Version: 6.8 Doc Build Date: 01/21/2020 Copyright © Trifacta Inc. 2020 - All Rights Reserved. CONFIDENTIAL These materials (the “Documentation”) are the confidential and proprietary information of Trifacta Inc. and may not be reproduced, modified, or distributed without the prior written permission of Trifacta Inc. EXCEPT AS OTHERWISE PROVIDED IN AN EXPRESS WRITTEN AGREEMENT, TRIFACTA INC. PROVIDES THIS DOCUMENTATION AS-IS AND WITHOUT WARRANTY AND TRIFACTA INC. DISCLAIMS ALL EXPRESS AND IMPLIED WARRANTIES TO THE EXTENT PERMITTED, INCLUDING WITHOUT LIMITATION THE IMPLIED WARRANTIES OF MERCHANTABILITY, NON-INFRINGEMENT AND FITNESS FOR A PARTICULAR PURPOSE AND UNDER NO CIRCUMSTANCES WILL TRIFACTA INC. BE LIABLE FOR ANY AMOUNT GREATER THAN ONE HUNDRED DOLLARS ($100) BASED ON ANY USE OF THE DOCUMENTATION. For third-party license information, please select About Trifacta from the Help menu. 1. Release Notes . 4 1.1 Changes to System Behavior . 4 1.1.1 Changes to the Language . 4 1.1.2 Changes to the APIs . 18 1.1.3 Changes to Configuration 23 1.1.4 Changes to the Object Model . 26 1.2 Release Notes 6.8 . 30 1.3 Release Notes 6.4 . 36 1.4 Release Notes 6.0 . 42 1.5 Release Notes 5.1 . 49 2. Quick Start 55 2.1 Install from AWS Marketplace with EMR . 55 2.2 Upgrade for AWS Marketplace with EMR . 62 3. Configure 62 3.1 Configure for AWS . 62 3.1.1 Configure for EC2 Role-Based Authentication . 68 3.1.2 Enable S3 Access . 70 3.1.2.1 Create Redshift Connections 81 3.1.3 Configure for EMR . -

The Platform Inside and out Release 0.8
The Platform Inside and Out Release 0.8 Joshua Patterson – GM, Data Science RAPIDS End-to-End Accelerated GPU Data Science Data Preparation Model Training Visualization Dask cuDF cuIO cuML cuGraph PyTorch Chainer MxNet cuXfilter <> pyViz Analytics Machine Learning Graph Analytics Deep Learning Visualization GPU Memory 2 Data Processing Evolution Faster data access, less data movement Hadoop Processing, Reading from disk HDFS HDFS HDFS HDFS HDFS Read Query Write Read ETL Write Read ML Train Spark In-Memory Processing 25-100x Improvement Less code HDFS Language flexible Read Query ETL ML Train Primarily In-Memory Traditional GPU Processing 5-10x Improvement More code HDFS GPU CPU GPU CPU GPU ML Language rigid Query ETL Read Read Write Read Write Read Train Substantially on GPU 3 Data Movement and Transformation The bane of productivity and performance APP B Read Data APP B GPU APP B Copy & Convert Data CPU GPU Copy & Convert Copy & Convert APP A GPU Data APP A Load Data APP A 4 Data Movement and Transformation What if we could keep data on the GPU? APP B Read Data APP B GPU APP B Copy & Convert Data CPU GPU Copy & Convert Copy & Convert APP A GPU Data APP A Load Data APP A 5 Learning from Apache Arrow ● Each system has its own internal memory format ● All systems utilize the same memory format ● 70-80% computation wasted on serialization and deserialization ● No overhead for cross-system communication ● Similar functionality implemented in multiple projects ● Projects can share functionality (eg, Parquet-to-Arrow reader) From Apache Arrow -

Arrow: Integration to 'Apache' 'Arrow'
Package ‘arrow’ September 5, 2021 Title Integration to 'Apache' 'Arrow' Version 5.0.0.2 Description 'Apache' 'Arrow' <https://arrow.apache.org/> is a cross-language development platform for in-memory data. It specifies a standardized language-independent columnar memory format for flat and hierarchical data, organized for efficient analytic operations on modern hardware. This package provides an interface to the 'Arrow C++' library. Depends R (>= 3.3) License Apache License (>= 2.0) URL https://github.com/apache/arrow/, https://arrow.apache.org/docs/r/ BugReports https://issues.apache.org/jira/projects/ARROW/issues Encoding UTF-8 Language en-US SystemRequirements C++11; for AWS S3 support on Linux, libcurl and openssl (optional) Biarch true Imports assertthat, bit64 (>= 0.9-7), methods, purrr, R6, rlang, stats, tidyselect, utils, vctrs RoxygenNote 7.1.1.9001 VignetteBuilder knitr Suggests decor, distro, dplyr, hms, knitr, lubridate, pkgload, reticulate, rmarkdown, stringi, stringr, testthat, tibble, withr Collate 'arrowExports.R' 'enums.R' 'arrow-package.R' 'type.R' 'array-data.R' 'arrow-datum.R' 'array.R' 'arrow-tabular.R' 'buffer.R' 'chunked-array.R' 'io.R' 'compression.R' 'scalar.R' 'compute.R' 'config.R' 'csv.R' 'dataset.R' 'dataset-factory.R' 'dataset-format.R' 'dataset-partition.R' 'dataset-scan.R' 'dataset-write.R' 'deprecated.R' 'dictionary.R' 'dplyr-arrange.R' 'dplyr-collect.R' 'dplyr-eval.R' 'dplyr-filter.R' 'expression.R' 'dplyr-functions.R' 1 2 R topics documented: 'dplyr-group-by.R' 'dplyr-mutate.R' 'dplyr-select.R' 'dplyr-summarize.R' -

In Reference to RPC: It's Time to Add Distributed Memory
In Reference to RPC: It’s Time to Add Distributed Memory Stephanie Wang, Benjamin Hindman, Ion Stoica UC Berkeley ACM Reference Format: D (e.g., Apache Spark) F (e.g., Distributed TF) Stephanie Wang, Benjamin Hindman, Ion Stoica. 2021. In Refer- A B C i ii 1 2 3 ence to RPC: It’s Time to Add Distributed Memory. In Workshop RPC E on Hot Topics in Operating Systems (HotOS ’21), May 31-June 2, application 2021, Ann Arbor, MI, USA. ACM, New York, NY, USA, 8 pages. Figure 1: A single “application” actually consists of many https://doi.org/10.1145/3458336.3465302 components and distinct frameworks. With no shared address space, data (squares) must be copied between 1 Introduction different components. RPC has been remarkably successful. Most distributed ap- Load balancer Scheduler + Mem Mgt plications built today use an RPC runtime such as gRPC [3] Executors or Apache Thrift [2]. The key behind RPC’s success is the Servers request simple but powerful semantics of its programming model. client data request cache In particular, RPC has no shared state: arguments and return Distributed object store values are passed by value between processes, meaning that (a) (b) they must be copied into the request or reply. Thus, argu- ments and return values are inherently immutable. These Figure 2: Logical RPC architecture: (a) today, and (b) with a shared address space and automatic memory management. simple semantics facilitate highly efficient and reliable imple- mentations, as no distributed coordination is required, while then return a reference, i.e., some metadata that acts as a remaining useful for a general set of distributed applications. -

Ray: a Distributed Framework for Emerging AI Applications
Ray: A Distributed Framework for Emerging AI Applications Philipp Moritz, Robert Nishihara, Stephanie Wang, Alexey Tumanov, Richard Liaw, Eric Liang, Melih Elibol, Zongheng Yang, William Paul, Michael I. Jordan, and Ion Stoica, UC Berkeley https://www.usenix.org/conference/osdi18/presentation/nishihara This paper is included in the Proceedings of the 13th USENIX Symposium on Operating Systems Design and Implementation (OSDI ’18). October 8–10, 2018 • Carlsbad, CA, USA ISBN 978-1-939133-08-3 Open access to the Proceedings of the 13th USENIX Symposium on Operating Systems Design and Implementation is sponsored by USENIX. Ray: A Distributed Framework for Emerging AI Applications Philipp Moritz,∗ Robert Nishihara,∗ Stephanie Wang, Alexey Tumanov, Richard Liaw, Eric Liang, Melih Elibol, Zongheng Yang, William Paul, Michael I. Jordan, Ion Stoica University of California, Berkeley Abstract and their use in prediction. These frameworks often lever- age specialized hardware (e.g., GPUs and TPUs), with the The next generation of AI applications will continuously goal of reducing training time in a batch setting. Examples interact with the environment and learn from these inter- include TensorFlow [7], MXNet [18], and PyTorch [46]. actions. These applications impose new and demanding The promise of AI is, however, far broader than classi- systems requirements, both in terms of performance and cal supervised learning. Emerging AI applications must flexibility. In this paper, we consider these requirements increasingly operate in dynamic environments, react to and present Ray—a distributed system to address them. changes in the environment, and take sequences of ac- Ray implements a unified interface that can express both tions to accomplish long-term goals [8, 43]. -

Zero-Cost, Arrow-Enabled Data Interface for Apache Spark
Zero-Cost, Arrow-Enabled Data Interface for Apache Spark Sebastiaan Jayjeet Aaron Chu Ivo Jimenez Jeff LeFevre Alvarez Chakraborty UC Santa Cruz UC Santa Cruz UC Santa Cruz Rodriguez UC Santa Cruz [email protected] [email protected] [email protected] Leiden University [email protected] [email protected] Carlos Alexandru Uta Maltzahn Leiden University UC Santa Cruz [email protected] [email protected] ABSTRACT which is a very expensive operation; (2) data processing sys- Distributed data processing ecosystems are widespread and tems require new adapters or readers for each new data type their components are highly specialized, such that efficient to support and for each new system to integrate with. interoperability is urgent. Recently, Apache Arrow was cho- A common example where these two issues occur is the sen by the community to serve as a format mediator, provid- de-facto standard data processing engine, Apache Spark. In ing efficient in-memory data representation. Arrow enables Spark, the common data representation passed between op- efficient data movement between data processing and storage erators is row-based [5]. Connecting Spark to other systems engines, significantly improving interoperability and overall such as MongoDB [8], Azure SQL [7], Snowflake [11], or performance. In this work, we design a new zero-cost data in- data sources such as Parquet [30] or ORC [3], entails build- teroperability layer between Apache Spark and Arrow-based ing connectors and converting data. Although Spark was data sources through the Arrow Dataset API. Our novel data initially designed as a computation engine, this data adapter interface helps separate the computation (Spark) and data ecosystem was necessary to enable new types of workloads. -

ECSEL2017-1-737451 Fitoptivis from the Cloud to the Edge
Ref. Ares(2019)3751938 - 12/06/2019 ECSEL2017-1-737451 FitOptiVis From the cloud to the edge - smart IntegraTion and OPtimisation Technologies for highly efficient Image and VIdeo processing Systems Deliverable: D5.1 - Components Analysis and Specification Due date of deliverable: (31-5-2019) Actual submission date: (11-06-2019) Start date of Project: 01 June 2018 Duration: 36 months Responsible: Zaid Al-Ars (Delft University of Technology) Revision: draft Dissemination level PU Public Restricted to other programme participants (including the Commission PP Service Restricted to a group specified by the consortium (including the RE Commission Services) Confidential, only for members of the consortium (excluding the CO Commission Services) WP5 D5.1, version 1.0 FitOptiVis ECSEL2017-1-737451 DOCUMENT INFO Author Author Company E-mail Francesca Palumbo UNISS [email protected] Carlo Sau, Tiziana UNICA [email protected]; Fanni, Luigi Raffo [email protected]; [email protected] Zaid Al-Ars TUDelft [email protected] Pablo Chaves SCHN [email protected] David Pampliega [email protected] Pablo Sánchez UC [email protected] Roman Čečil UWB [email protected] Document history Document Date Change version # V0.1 25/01/2019 Added table of content and initial inputs from survey V0.2 07/05/2019 Integrated partner contributions from Feb to Apr V0.3 14/05/2019 Integrated partner contributions till May 14 V0.4 22/05/2019 Integrated partner contributions till Brno meeting V0.5 29/05/2019 Integrated partner contributions till May 29 -
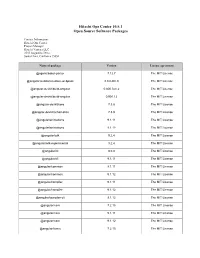
Open Source Software Packages
Hitachi Ops Center 10.5.1 Open Source Software Packages Contact Information: Hitachi Ops Center Project Manager Hitachi Vantara LLC 2535 Augustine Drive Santa Clara, California 95054 Name of package Version License agreement @agoric/babel-parser 7.12.7 The MIT License @angular-builders/custom-webpack 8.0.0-RC.0 The MIT License @angular-devkit/build-angular 0.800.0-rc.2 The MIT License @angular-devkit/build-angular 0.901.12 The MIT License @angular-devkit/core 7.3.8 The MIT License @angular-devkit/schematics 7.3.8 The MIT License @angular/animations 9.1.11 The MIT License @angular/animations 9.1.12 The MIT License @angular/cdk 9.2.4 The MIT License @angular/cdk-experimental 9.2.4 The MIT License @angular/cli 8.0.0 The MIT License @angular/cli 9.1.11 The MIT License @angular/common 9.1.11 The MIT License @angular/common 9.1.12 The MIT License @angular/compiler 9.1.11 The MIT License @angular/compiler 9.1.12 The MIT License @angular/compiler-cli 9.1.12 The MIT License @angular/core 7.2.15 The MIT License @angular/core 9.1.11 The MIT License @angular/core 9.1.12 The MIT License @angular/forms 7.2.15 The MIT License @angular/forms 9.1.0-next.3 The MIT License @angular/forms 9.1.11 The MIT License @angular/forms 9.1.12 The MIT License @angular/language-service 9.1.12 The MIT License @angular/platform-browser 7.2.15 The MIT License @angular/platform-browser 9.1.11 The MIT License @angular/platform-browser 9.1.12 The MIT License @angular/platform-browser-dynamic 7.2.15 The MIT License @angular/platform-browser-dynamic 9.1.11 The MIT License @angular/platform-browser-dynamic -

Enabling Geospatial in Big Data Lakes and Databases with Locationtech Geomesa
Enabling geospatial in big data lakes and databases with LocationTech GeoMesa ApacheCon@Home 2020 James Hughes James Hughes ● CCRi’s Director of Open Source Programs ● Working in geospatial software on the JVM for the last 8 years ● GeoMesa core committer / product owner ● SFCurve project lead ● JTS committer ● Contributor to GeoTools and GeoServer ● What type? Big Geospatial Data ● What volume? Problem Statement for today: Problem: How do we handle “big” geospatial data? Problem Statement for today: Problem: How do we handle “big” geospatial data? First refinement: What type of data do are we interested in? Vector Raster Point Cloud Problem Statement for today: Problem: How do we handle “big” geospatial data? First refinement: What type of data do are we interested in? Vector Raster Point Cloud Problem Statement for today: Problem: How do we handle “big” vector geospatial data? Second refinement: How much data is “big”? What is an example? GDELT: Global Database of Event, Language, and Tone “The GDELT Event Database records over 300 categories of physical activities around the world, from riots and protests to peace appeals and diplomatic exchanges, georeferenced to the city or mountaintop, across the entire planet dating back to January 1, 1979 and updated every 15 minutes.” ~225-250 million records Problem Statement for today: Problem: How do we handle “big” vector geospatial data? Second refinement: How much data is “big”? What is an example? Open Street Map: OpenStreetMap is a collaborative project to create a free editable map of the world. The geodata underlying the map is considered the primary output of the project. -

Release Notes Date Published: 2020-10-13 Date Modified
Cloudera Runtime 7.1.4 Release Notes Date published: 2020-10-13 Date modified: https://docs.cloudera.com/ Legal Notice © Cloudera Inc. 2021. All rights reserved. The documentation is and contains Cloudera proprietary information protected by copyright and other intellectual property rights. No license under copyright or any other intellectual property right is granted herein. Copyright information for Cloudera software may be found within the documentation accompanying each component in a particular release. Cloudera software includes software from various open source or other third party projects, and may be released under the Apache Software License 2.0 (“ASLv2”), the Affero General Public License version 3 (AGPLv3), or other license terms. Other software included may be released under the terms of alternative open source licenses. Please review the license and notice files accompanying the software for additional licensing information. Please visit the Cloudera software product page for more information on Cloudera software. For more information on Cloudera support services, please visit either the Support or Sales page. Feel free to contact us directly to discuss your specific needs. Cloudera reserves the right to change any products at any time, and without notice. Cloudera assumes no responsibility nor liability arising from the use of products, except as expressly agreed to in writing by Cloudera. Cloudera, Cloudera Altus, HUE, Impala, Cloudera Impala, and other Cloudera marks are registered or unregistered trademarks in the United States and other countries. All other trademarks are the property of their respective owners. Disclaimer: EXCEPT AS EXPRESSLY PROVIDED IN A WRITTEN AGREEMENT WITH CLOUDERA, CLOUDERA DOES NOT MAKE NOR GIVE ANY REPRESENTATION, WARRANTY, NOR COVENANT OF ANY KIND, WHETHER EXPRESS OR IMPLIED, IN CONNECTION WITH CLOUDERA TECHNOLOGY OR RELATED SUPPORT PROVIDED IN CONNECTION THEREWITH. -

Processingではじめるプログラミング 楽しく、簡単に、Javaベースの言語を学ぶ (I/O)/加藤直樹/ 著 IO編集部/編集/NEOB
Processingではじめるプログラミング 楽しく、簡単に、Javaベースの言語を学ぶ - ダウ ンロード, PDF オンラインで読む ダウンロード オンラインで読む 概要 「Processing」の開発環境の導入から、「図形を描く」「色を塗る」といった基礎、「お絵かきスケッ チ」「算数教材」な 朝倉書店 (A5) 【2007年10月発売】 ISBNコード 9784254116366. 価格:3,132円(本体:2,900円 +税). 在庫状況:在庫あり(1~2日で出荷). 買い物かごに入れる · 欲しいものリストに追加する. Processingではじめるプログラミング. 楽しく、簡単に、Javaベースの言語を学ぶ I/O books. 加藤直樹. 工学社 (A5) 【2013年02月発売】 ISBNコード 9784777517428. 価格:2,484円(本 体:2,300円+税). 在庫状況:品切れのため入荷お知らせにご登録下さい. 入荷お知らせ希望に 登録 · 欲しいものリストに追加する · 熱力学. 教科書:清水忠昭・菅田一博 『新・ C 言語のススメ -C で始めるプログラミング-』 (サイエンス 社,2006) . また実際にそれらの機能を応用したプログラムの作成を UNIX システム上で実習する。 授業内容・授業計画. 以下の内容について講義と演習を交互に繰り返しながら単元を進めていく。 1. 概要説明・ウォームアップ. 2. 簡単なプログラミング. 3. 変数、代入. 4. 入力処理. 5. 条件分岐. 6. ... また、大学の「微分積分」が同時並行に進められるため、この講座を学ぶことが、予習復習も 兼ねることになる。 授業内容・授業計画. 2017年11月27日 . http://shiffman.net/ Daniel Shiffman - Wikipedia ダニエル・シフマンは ProcessingというJavaベースのグラフィックス開発のプログラミング言語を作っている財団のリード・デベ ロッパーで芸術系の学校の准教授だそうだ。 . 彼はThe Coding Trainというプログラミングを学べる 動画をYouTube上に公開している。 . 初心者でも随分と簡単に画像を操作するプログラムが作れ るので、円などを動かしたり、表を作ったりするのが簡単で初心者でも最初から視覚的なプログラム を作って学ぶことができる。 言語実装の楽しさ、どのように動いているかについてを共有をしたいと考えております。 cafebabepyの cafebabeとはJavaクラスファイルのマジックナンバーです。 pyはPythonのpyです。 JVM上で動く Jythonは公式 . 新しいプログラミング言語の学び方 ~ HTTPサーバーを作って学ぶJava, Scala, Clojure ~. 田所 駿佑 .. 社内新規プロダクトでDDD, CQRSの思想をベースとしたアーキテクチャを 構築し、コマンド(更新系処理)ではSpring Data JPA(Hibernate)を、クエリ(参照系処理)ではjOOQ を採用しました。結果として. 多言語、多通貨、複数会計基準、. マルチカンパニー管理に対応した「A.S.I.A.GP」を現地法人の 業務基盤として利用することで、これまでに把握が. 難しかった海外現地法人の経営状況をリアルタ イムに把握することができます。 製品 / ソリューション説明と特徴: .. を割り当て、作業を管理できま す。また、コメント機能によりチーム内でディスカッションし、内容を履歴として. 残すことができます。ま たメンション機能で特定の担当者に簡単に情報の共有ができます。JIRA -

High-Throughput Big Data Analytics Through Accelerated Parquet to Arrow Conversion
High-Throughput Big Data Analytics Through Accelerated Parquet to Arrow Conversion L.T.J. van Leeuwen High-Throughput Big Data Analytics Through Accelerated Parquet to Arrow Conversion ATHESIS submitted in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE in COMPUTER ENGINEERING and EMBEDDED SYSTEMS by Laurentius Theodorus Johannes van Leeuwen To be defended on Tuesday the 27th of August COMMITTEE: Q&CE-CE-MS-2019-17 Chair: by L.T.J. van Leeuwen (4239784) Dr. ir. Z. Al-Ars Faculty of EEMCS Delft University of Technology Members: Mekelweg 4 Prof. dr. H.P. Hofstee Delft, The Netherlands Dr. J.S. Rellermeyer Ir. J.W. Peltenburg i Abstract With the advent of high-bandwidth non-volatile storage devices, the classical assumption that database analytics applications are bottlenecked by CPUs having to wait for slow I/O devices is being flipped around. Instead, CPUs are no longer able to decompress and deseri- alize the data stored in storage-focused file formats fast enough to keep up with the speed at which compressed data is read from storage. In order to better utilize the increasing I/O band- width, this work proposes a hardware accelerated approach to converting storage-focused file formats to in-memory data structures. To that end, an FPGA-based Apache Parquet reading en- gine is developed that utilizes existing FPGA and memory interfacing hardware to write data to memory in Apache Arrow’s in-memory format. A modular and expandable hardware archi- tecture called the ParquetReader with out-of-the-box support for DELTA_BINARY_PACKED and DELTA_LENGTH_BYTE_ARRAY encodings is proposed and implemented on an Amazon EC2 F1 instance with an XCVU9P FPGA.