Heliconius Butterflies Host Characteristic and Phylogenetically Structured Adult-Stage Microbiomes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lizard Predation on Tropical Butterflies
Journal of the Lepidopterists' Societ!J 36(2), 1982, 148-152 LIZARD PREDATION ON TROPICAL BUTTERFLIES PAUL R EHRLICH AND ANNE H, EHRLICH Deparhnent of Biological Sciences, Stanford University, Stanford, California 94305 ABSTRACT. Iguanid lizards at Iguac;u Falls, Brazil appear to make butterflies a major component of their diets. They both stalk sitting individuals and leap into the air to capture ones in flight. Butterfly species seem to be attacked differentially. These observations support the widespread assumption that lizards can be involved as selec tive agents in the evolution of butterfly color patterns and behavior. Butterflies have been prominent in the development of ideas about protective and warning coloration and mimicry (e.g., Cott, 1940; J. Brower, 1958; M. Rothschild, 1972), and the dynamics of natural pop ulations (Ford & Ford, 1930; Ehrlich et aI., 1975). In spite of the crucial role that predation on adults must play in evolution of defen sive coloration and may play in population dynamics, there is re markably little information on predation on adult butterflies in nature. This lack is all the more striking, considering the large numbers of people who collect butterflies and the abundant indirect evidence from bird beak and lizard jaw marks on butterfly wings (e.g., Carpen ter, 1937; Shapiro, 1974) that adult butterflies are quite frequently attacked. Published field observations of predation on butterflies deal almost exclusively with the attacks of birds and consist largely of accounts of individual attacks (Fryer, 1913). Observations of natural predation by lizards are very rare, although "birds and lizards have long been considered to be the major selective agents responsible for the ex treme diversity of unpalatable and mimetic forms of butterflies in nature" (Boyden, 1976). -

Alfred Russel Wallace and the Darwinian Species Concept
Gayana 73(2): Suplemento, 2009 ISSN 0717-652X ALFRED RUSSEL WALLACE AND THE Darwinian SPECIES CONCEPT: HIS paper ON THE swallowtail BUTTERFLIES (PAPILIONIDAE) OF 1865 ALFRED RUSSEL WALLACE Y EL concepto darwiniano DE ESPECIE: SU TRABAJO DE 1865 SOBRE MARIPOSAS papilio (PAPILIONIDAE) Jam ES MA LLET 1 Galton Laboratory, Department of Biology, University College London, 4 Stephenson Way, London UK, NW1 2HE E-mail: [email protected] Abstract Soon after his return from the Malay Archipelago, Alfred Russel Wallace published one of his most significant papers. The paper used butterflies of the family Papilionidae as a model system for testing evolutionary hypotheses, and included a revision of the Papilionidae of the region, as well as the description of some 20 new species. Wallace argued that the Papilionidae were the most advanced butterflies, against some of his colleagues such as Bates and Trimen who had claimed that the Nymphalidae were more advanced because of their possession of vestigial forelegs. In a very important section, Wallace laid out what is perhaps the clearest Darwinist definition of the differences between species, geographic subspecies, and local ‘varieties.’ He also discussed the relationship of these taxonomic categories to what is now termed ‘reproductive isolation.’ While accepting reproductive isolation as a cause of species, he rejected it as a definition. Instead, species were recognized as forms that overlap spatially and lack intermediates. However, this morphological distinctness argument breaks down for discrete polymorphisms, and Wallace clearly emphasised the conspecificity of non-mimetic males and female Batesian mimetic morphs in Papilio polytes, and also in P. -

Title Lorem Ipsum Dolor Sit Amet, Consectetur Adipiscing Elit
Volume 26: 102–108 METAMORPHOSIS www.metamorphosis.org.za ISSN 1018–6490 (PRINT) LEPIDOPTERISTS’ SOCIETY OF AFRICA ISSN 2307–5031 (ONLINE) Classification of the Afrotropical butterflies to generic level Published online: 25 December 2015 Mark C. Williams 183 van der Merwe Street, Rietondale, Pretoria, South Africa. E-mail: [email protected] Copyright © Lepidopterists’ Society of Africa Abstract: This paper applies the findings of phylogenetic studies on butterflies (Papilionoidea) in order to present an up to date classification of the Afrotropical butterflies to genus level. The classification for Afrotropical butterflies is placed within a worldwide context to subtribal level. Taxa that still require interrogation are highlighted. Hopefully this classification will provide a stable context for researchers working on Afrotropical butterflies. Key words: Lepidoptera, Papilionoidea, Afrotropical butterflies, classification. Citation: Williams, M.C. (2015). Classification of the Afrotropical butterflies to generic level. Metamorphosis 26: 102–108. INTRODUCTION Suborder Glossata Fabricius, 1775 (6 infraorders) Infraorder Heteroneura Tillyard, 1918 (34 Natural classifications of biological organisms, based superfamilies) on robust phylogenetic hypotheses, are needed before Clade Obtectomera Minet, 1986 (12 superfamilies) meaningful studies can be conducted in regard to their Superfamily Papilionoidea Latreille, 1802 (7 evolution, biogeography, ecology and conservation. families) Classifications, dating from the time of Linnaeus in the Family Papilionidae Latreille, 1802 (32 genera, 570 mid seventeen hundreds, were based on morphology species) for nearly two hundred and fifty years. Classifications Family Hedylidae Guenée, 1858 (1 genus, 36 species) based on phylogenies derived from an interrogation of Family Hesperiidae Latreille, 1809 (570 genera, 4113 the genome of individual organisms began in the late species) 20th century. -

Distinction of Grapholita Molesta Busck and Grapholita Dimorpha Komai Larvae Based on Morphological Feature of Anal Prolegs
九州大学学術情報リポジトリ Kyushu University Institutional Repository Distinction of Grapholita molesta Busck and Grapholita dimorpha Komai larvae based on morphological feature of anal prolegs Lee, Seung–Yeol College of Agriculture and Life Sciences, Kyungpook National University Choi, Kwang–Shik College of Natural Sciences, Kyungpook National University Back, Chang–Gi National Institute of Horticultural and Herbal Science, Rural Development Administration Choi, Kyung–Hee National Institute of Horticultural and Herbal Science, Rural Development Administration 他 https://doi.org/10.5109/1526340 出版情報:九州大学大学院農学研究院紀要. 60 (2), pp.291-295, 2015-09-18. 九州大学大学院農学研究 院 バージョン: 権利関係: J. Fac. Agr., Kyushu Univ., 60 (2), 291–295 (2015) Distinction of Grapholita molesta Busck and Grapholita dimorpha Komai larvae based on morphological feature of anal prolegs Seung–Yeol LEE1, Kwang–Shik CHOI2, Chang–Gi BACK3, Kyung–Hee CHOI3, In–Kyu KANG1, Hee–Young JUNG1* and Shoji OHGA* Laboratory of Forest Resources Management, Division of Forest Environmental Sciences, Department of Agro–environmental Sciences, Faculty of Agriculture, Kyushu University, Sasaguri, Fukuoka 811–2415, Japan (Received April 14, 2015 and accepted May 19, 2015) Larvae of Grapholita molesta Busck and Grapholita dimorpha Komai, which are major moth pests that affect apples in Korea, are very difficult to identify because of their morphological similarities. In this study, we investigated how to distinguish the larvae of these two species by using specific morphological features. Between 2013 and 2014, a total of 84 specimens were collected from apples suspected of infestation in Gunwi–gun and Cheongsong–gun, Gyeongsangbuk–do, Korea, and they were observed using a stereo microscope, optical microscope, and scanning electron microscope. -

The Genetics and Evolution of Iridescent Structural Colour in Heliconius Butterflies
The genetics and evolution of iridescent structural colour in Heliconius butterflies Melanie N. Brien A thesis submitted in partial fulfilment of the requirements for the degree of Doctor of Philosophy The University of Sheffield Faculty of Science Department of Animal & Plant Sciences Submission Date August 2019 1 2 Abstract The study of colouration has been essential in developing key concepts in evolutionary biology. The Heliconius butterflies are well-studied for their diverse aposematic and mimetic colour patterns, and these pigment colour patterns are largely controlled by a small number of homologous genes. Some Heliconius species also produce bright, highly reflective structural colours, but unlike pigment colour, little is known about the genetic basis of structural colouration in any species. In this thesis, I aim to explore the genetic basis of iridescent structural colour in two mimetic species, and investigate its adaptive function. Using experimental crosses between iridescent and non-iridescent subspecies of Heliconius erato and Heliconius melpomene, I show that iridescent colour is a quantitative trait by measuring colour variation in offspring. I then use a Quantitative Trait Locus (QTL) mapping approach to identify loci controlling the trait in the co-mimics, finding that the genetic basis is not the same in the two species. In H. erato, the colour is strongly sex-linked, while in H. melpomene, we find a large effect locus on chromosome 3, plus a number of putative small effect loci in each species. Therefore, iridescence in Heliconius is not an example of repeated gene reuse. I then show that both iridescent colour and pigment colour are sexually dimorphic in H. -

Evolution of Insect Color Vision: from Spectral Sensitivity to Visual Ecology
EN66CH23_vanderKooi ARjats.cls September 16, 2020 15:11 Annual Review of Entomology Evolution of Insect Color Vision: From Spectral Sensitivity to Visual Ecology Casper J. van der Kooi,1 Doekele G. Stavenga,1 Kentaro Arikawa,2 Gregor Belušic,ˇ 3 and Almut Kelber4 1Faculty of Science and Engineering, University of Groningen, 9700 Groningen, The Netherlands; email: [email protected] 2Department of Evolutionary Studies of Biosystems, SOKENDAI Graduate University for Advanced Studies, Kanagawa 240-0193, Japan 3Department of Biology, Biotechnical Faculty, University of Ljubljana, 1000 Ljubljana, Slovenia; email: [email protected] 4Lund Vision Group, Department of Biology, University of Lund, 22362 Lund, Sweden; email: [email protected] Annu. Rev. Entomol. 2021. 66:23.1–23.28 Keywords The Annual Review of Entomology is online at photoreceptor, compound eye, pigment, visual pigment, behavior, opsin, ento.annualreviews.org anatomy https://doi.org/10.1146/annurev-ento-061720- 071644 Abstract Annu. Rev. Entomol. 2021.66. Downloaded from www.annualreviews.org Copyright © 2021 by Annual Reviews. Color vision is widespread among insects but varies among species, depend- All rights reserved ing on the spectral sensitivities and interplay of the participating photore- Access provided by University of New South Wales on 09/26/20. For personal use only. ceptors. The spectral sensitivity of a photoreceptor is principally determined by the absorption spectrum of the expressed visual pigment, but it can be modified by various optical and electrophysiological factors. For example, screening and filtering pigments, rhabdom waveguide properties, retinal structure, and neural processing all influence the perceived color signal. -

Region of Nymphalidae and Libytheidae (Lepidoptera) from Japan
MORPHOLOGICAL STUDIES ON THE OCCIPITAL REGION OF NYMPHALIDAE AND LIBYTHEIDAE (LEPIDOPTERA) FROM JAPAN Takashi TSUBUKI and Nagao KOYAMA * .h2monji junior High School, 10-33, Kita6tsz{ha 1, Toshima-feu, Toleyo (170), 1opan ** Biological Laboratory, Shinshi2 Udeiversity, Cllada (386), 14pan CONTENTS I. Introduction・--・ny・・・・・・・・・・・・・・・・・-・-・・・・・・-・---・--t・-・---・ny・・・・-・-・・・-・-・・-・・・1 II. Materials and methods・-・・・・・・・・・t・・・・・・t・・・・・・・・・・・・・・`・・・・・・・・・・-・・・-・・・・・・-d・・・・・・・・・・・・2 III. General morphology of the occipital region ・・・・・・}・・・・・・・・・・・・・・・・・・・・・・・・・・・・・4 IV. Occipital structure of each species in Nymphaliclae and Libytheidae ・-ib-・・・・-・・・-・ny・・・・・--・-・・・・・・・-・・-・----・b-・・・----・-・・・-・・-・--・・-6 V. Phylogenical grouping of Nymphalidae based on the occipital structure・t・・・・・・・--・--・・-・-・・・・・・・・・・-・・・・-ny-・・・ny-・--・-----・H"""""".","".."."lg VI. Summary ・・・・-・-・-・・・・・・-・・----・・・-・・・・-・-・-・-----・・-------・-・----・23 VII. Literatures cited ・・・--・・・・・・・・・`・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・--・-・・-・・・・・・tt・・・・・・23 I. INTRODUCTION A considerable number of studies have been carried out on the occipital morphology of moths since YAGI and KoYAMA's report of 1963 (KoYAMA and MlyATA, 1969, 1970, 1975;MIYATA, 1971, 1972, 1973, 1974, 1975;MIyATA and KOYAMA, 1971, 1972, 1976). In the butterfiies, however, few papers were available concerning the occipital region, on which EHRucH (1958, a b), YAGI and KoyAMA (1963) and KoyAMA and OGAwA (1972) briefly described. Then, tried to study the occipital region of butterflies, the authors observed it preliminarily (TsuBuKI et aL 1975). The present paper deals with the occipital structure of Nymphalidae and Libytheidae from Japan with reference to its bearing on systematics. Before going further, the authors wish to express their gratitude to Prof. Dr. H. SAwADA and Prof. Dr. N. GOKAN, Tokyo University of Agriculture, for their advice and assistance. Thanks are also due to Mr. N. K6DA who gave valuable materials for this work, and to Mr. Y. YAGucm and the members of JEOL Ltd. -
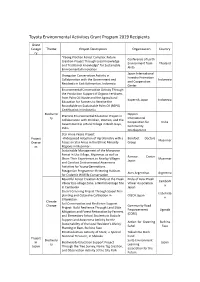
List of Previous Grant Projects
Toyota Environmental Activities Grant Program 2019 Recipients Grant Catego Theme Project Description Organization Country ry "Kaeng Krachan Forest Complex: Future Conference of Earth Creation Project Through Local Knowledge Environment from Thailand and Traditional Knowledge" for Sustainable Akita Environmental Innovation Japan International Orangutan Conservation Activity in Forestry Promotion Collaboration with the Government and Indonesia and Cooperation Residents in East Kalimantan, Indonesia Center Environmental Conservation Activity Through the Production Support of Organic Fertilizers from Palm Oil Waste and the Agricultural Kopernik Japan Indonesia Education for Farmers to Receive the Roundtable on Sustainable Palm Oil (RSPO) Certification in Indonesia Biodiversi Nippon Practical Environmental Education Project in ty International Collaboration with Children, Women, and the Cooperation for India Government in a Rural Village in Bodh Gaya, Community India Development Star Anise Peace Project Project -Widespread Adoption of Agroforestry with a Barefoot Doctors Myanmar Overse Focus on Star Anise in the Ethnic Minority Group as Regions in Myanmar- Sustainable Management of the Mangrove Forest in Uto Village, Myanmar, as well as Ramsar Center Share Their Experiences to Nearby Villages Myanmar Japan and Conduct Environmental Awareness Activities for Young Generations Patagonian Programme: Restoring Habitats Aves Argentinas Argentina for Endemic Wildlife Conservation Beautiful Forest Creation Activity at the Preah Pride of Asia: Preah -

The Speciation History of Heliconius: Inferences from Multilocus DNA Sequence Data
The speciation history of Heliconius: inferences from multilocus DNA sequence data by Margarita Sofia Beltrán A thesis submitted for the degree of Doctor of Philosophy of the University of London September 2004 Department of Biology University College London 1 Abstract Heliconius butterflies, which contain many intermediate stages between local varieties, geographic races, and sympatric species, provide an excellent biological model to study evolution at the species boundary. Heliconius butterflies are warningly coloured and mimetic, and it has been shown that these traits can act as a form of reproductive isolation. I present a species-level phylogeny for this group based on 3834bp of mtDNA (COI, COII, 16S) and nuclear loci (Ef1α, dpp, ap, wg). Using these data I test the geographic mode of speciation in Heliconius and whether mimicry could drive speciation. I found little evidence for allopatric speciation. There are frequent shifts in colour pattern within and between sister species which have a positive and significant correlation with species diversity; this suggests that speciation is facilitated by the evolution of novel mimetic patterns. My data is also consistent with the idea that two major innovations in Heliconius, adult pollen feeding and pupal-mating, each evolved only once. By comparing gene genealogies from mtDNA and introns from nuclear Tpi and Mpi genes, I investigate recent speciation in two sister species pairs, H. erato/H. himera and H. melpomene/H. cydno. There is highly significant discordance between genealogies of the three loci, which suggests recent speciation with ongoing gene flow. Finally, I explore the phylogenetic relationships between races of H. melpomene using an AFLP band tightly linked to the Yb colour pattern locus (which determines the yellow bar in the hindwing). -

Genomic Architecture of Adaptive Color Pattern Divergence and Convergence in Heliconius Butterflies
Downloaded from genome.cshlp.org on September 29, 2021 - Published by Cold Spring Harbor Laboratory Press Research Genomic architecture of adaptive color pattern divergence and convergence in Heliconius butterflies Megan A. Supple,1,2 Heather M. Hines,3,4 Kanchon K. Dasmahapatra,5,6 James J. Lewis,7 Dahlia M. Nielsen,3 Christine Lavoie,8 David A. Ray,8 Camilo Salazar,1,9 W. Owen McMillan,1,10 and Brian A. Counterman8,10,11 1Smithsonian Tropical Research Institute, Panama City, Republic of Panama; 2Biomathematics Program, North Carolina State University, Raleigh, North Carolina 27695, USA; 3Department of Genetics, North Carolina State University, Raleigh, North Carolina 27695, USA; 4Department of Biology, Pennsylvania State University, University Park, Pennsylvania 16802, USA; 5Department of Genetics, Evolution and Environment, University College London, London WC1E 6BT, United Kingdom; 6Department of Biology, University of York, York YO10 5DD, United Kingdom; 7Department of Ecology and Evolutionary Biology, Cornell University, Ithaca, New York 14853, USA; 8Department of Biological Sciences, Mississippi State University, Mississippi State, Mississippi 39762, USA; 9Facultad de Ciencias Naturales y Matema´ticas, Universidad del Rosario, Bogota´ DC, Colombia Identifying the genetic changes driving adaptive variation in natural populations is key to understanding the origins of biodiversity. The mosaic of mimetic wing patterns in Heliconius butterflies makes an excellent system for exploring adaptive variation using next-generation sequencing. In this study, we use a combination of techniques to annotate the genomic interval modulating red color pattern variation, identify a narrow region responsible for adaptive divergence and con- vergence in Heliconius wing color patterns, and explore the evolutionary history of these adaptive alleles. -

Photonic Crystal Structure of Wing Scales in Sasakia Charonda Butterflies
Materials Transactions, Vol. 51, No. 2 (2010) pp. 202 to 208 Special Issue on Development and Fabrication of Advanced Materials Assisted by Nanotechnology and Microanalysis #2010 The Japan Institute of Metals Photonic Crystal Structure of Wing Scales in Sasakia Charonda Butterflies Jirˇina Mateˇjkova´-Plsˇkova´1, Dalibor Jancˇik1, Miroslav Masˇla´nˇ1, Satoshi Shiojiri2 and Makoto Shiojiri3;* 1Centre for Nanomaterial Research, Faculty of Science, Palacky University in Olomouc, Slechtitelu 11, 783 71 Olomouc, Czech Republic 2Matsubara Junior High-school, Kyoto 604-8812, Japan 3Professor Emeritus of Kyoto Institute of Technology, 1-297 Wakiyama, Kyoto 618-0091, Japan The hindwings of the male Sasakia charonda charonda butterflies comprise iridescent purple-blue areas, iridescent white-pearl areas, yellow spots and red spots as well as brown background. We have examined the microstructure of their scales by scanning electron microscopy, for applying their photonic crystal structures to fine light manipulators such as reflection elements in laser diodes. The scales in the yellow spots, red spots and brown background have almost the same structure, which is an optical diffraction grating made of ridges with two cuticle layers. Their difference comes from the contained pigments. The scales in the iridescent purple-blue and white-pearl are also the same in structure. They have seven tilted cuticle layers lapped on the ridges, which also constitute a grating. The widths of the ridge and groove in the grating are different between scales of the two kinds. It is shown that the vivid iridescence is mainly attributed to multiple interferences caused between rays reflected from the seven cuticle layers with air gaps. -

Genetic Differentiation Without Mimicry Shift in a Pair of Hybridizing Heliconius Species (Lepidoptera: Nymphalidae)
bs_bs_banner Biological Journal of the Linnean Society, 2013, 109, 830–847. With 5 figures Genetic differentiation without mimicry shift in a pair of hybridizing Heliconius species (Lepidoptera: Nymphalidae) CLAIRE MÉROT1, JESÚS MAVÁREZ2,3, ALLOWEN EVIN4,5, KANCHON K. DASMAHAPATRA6,7, JAMES MALLET7,8, GERARDO LAMAS9 and MATHIEU JORON1* 1UMR CNRS 7205, Muséum National d’Histoire Naturelle, 45 rue Buffon, 75005 Paris, France 2LECA, BP 53, Université Joseph Fourier, 2233 Rue de la Piscine, 38041 Grenoble Cedex, France 3Smithsonian Tropical Research Institute, Apartado 2072, Balboa, Panama 4Department of Archaeology, University of Aberdeen, St. Mary’s Building, Elphinstone Road, Aberdeen AB24 3UF, UK 5UMR CNRS 7209, Muséum National d’Histoire Naturelle, 55 rue Buffon, 75005 Paris, France 6Department of Biology, University of York, Wentworth Way, Heslington, York YO10 5DD, UK 7Department of Genetics, Evolution and Environment, University College London, Gower Street, London WC1E 6BT, UK 8Department of Organismic and Evolutionary Biology, Harvard University, 16 Divinity Avenue, Cambridge, MA 02138, USA 9Museo de Historia Natural, Universidad Nacional Mayor San Marcos, Av. Arenales, 1256, Apartado 14-0434, Lima-14, Peru Received 22 January 2013; revised 25 February 2013; accepted for publication 25 February 2013 Butterflies in the genus Heliconius have undergone rapid adaptive radiation for warning patterns and mimicry, and are excellent models to study the mechanisms underlying diversification. In Heliconius, mimicry rings typically involve distantly related species, whereas closely related species often join different mimicry rings. Genetic and behavioural studies have shown how reproductive isolation in many pairs of Heliconius taxa is largely mediated by natural and sexual selection on wing colour patterns.