Jimmunol.1901059.Full.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Rare Modifier Variants Alter the Severity of Cardiovascular Disease in Pseudoxanthoma Elasticum
fcell-09-612581 June 8, 2021 Time: 11:34 # 1 BRIEF RESEARCH REPORT published: 08 June 2021 doi: 10.3389/fcell.2021.612581 Rare Modifier Variants Alter the Severity of Cardiovascular Disease in Pseudoxanthoma Elasticum: Identification of Novel Candidate Modifier Genes and Disease Pathways Through Mixture of Effects Analysis Eva Y. G. De Vilder1,2,3, Ludovic Martin4, Georges Lefthériotis5, Paul Coucke1,6, Filip Van Nieuwerburgh7 and Olivier M. Vanakker1,6* Edited by: 1 2 Svetlana Komarova, Center for Medical Genetics, Ghent University Hospital, Ghent, Belgium, The Research Foundation – Flanders, Ghent, 3 4 McGill University, Canada Belgium, Department of Ophthalmology, Ghent University Hospital, Ghent, Belgium, Department of Dermatology, Angers University Hospital, Angers, France, 5 Department of Vascular Physiology and Sports Medicine, Angers University, Angers, Reviewed by: France, 6 Department of Biomolecular Medicine, Ghent University, Ghent, Belgium, 7 Department of Pharmaceutics, Carolina Beraldo Meloto, Laboratory of Pharmaceutical Biotechnology, Ghent University, Ghent, Belgium McGill University, Canada Koen van de Wetering, Thomas Jefferson University, Introduction: Pseudoxanthoma elasticum (PXE), an ectopic mineralization disorder United States caused by pathogenic ABCC6 variants, is characterized by skin, ocular and Jessica Bertrand, University Hospital Magdeburg, cardiovascular (CV) symptoms. Due to striking phenotypic variability without genotype- Germany phenotype correlations, modifier genes are thought to play a role in disease variability. *Correspondence: In this study, we evaluated the collective modifying effect of rare variants on the Olivier M. Vanakker [email protected] cardiovascular phenotype of PXE. Materials and Methods: Mixed effects of rare variants were assessed by Whole Exome Specialty section: This article was submitted to Sequencing in 11 PXE patients with an extreme CV phenotype (mild/severe). -

Genetic Variation Across the Human Olfactory Receptor Repertoire Alters Odor Perception
bioRxiv preprint doi: https://doi.org/10.1101/212431; this version posted November 1, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Genetic variation across the human olfactory receptor repertoire alters odor perception Casey Trimmer1,*, Andreas Keller2, Nicolle R. Murphy1, Lindsey L. Snyder1, Jason R. Willer3, Maira Nagai4,5, Nicholas Katsanis3, Leslie B. Vosshall2,6,7, Hiroaki Matsunami4,8, and Joel D. Mainland1,9 1Monell Chemical Senses Center, Philadelphia, Pennsylvania, USA 2Laboratory of Neurogenetics and Behavior, The Rockefeller University, New York, New York, USA 3Center for Human Disease Modeling, Duke University Medical Center, Durham, North Carolina, USA 4Department of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, North Carolina, USA 5Department of Biochemistry, University of Sao Paulo, Sao Paulo, Brazil 6Howard Hughes Medical Institute, New York, New York, USA 7Kavli Neural Systems Institute, New York, New York, USA 8Department of Neurobiology and Duke Institute for Brain Sciences, Duke University Medical Center, Durham, North Carolina, USA 9Department of Neuroscience, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA *[email protected] ABSTRACT The human olfactory receptor repertoire is characterized by an abundance of genetic variation that affects receptor response, but the perceptual effects of this variation are unclear. To address this issue, we sequenced the OR repertoire in 332 individuals and examined the relationship between genetic variation and 276 olfactory phenotypes, including the perceived intensity and pleasantness of 68 odorants at two concentrations, detection thresholds of three odorants, and general olfactory acuity. -

OR2G2 (Human) Recombinant Protein (P01)
Produktinformation Diagnostik & molekulare Diagnostik Laborgeräte & Service Zellkultur & Verbrauchsmaterial Forschungsprodukte & Biochemikalien Weitere Information auf den folgenden Seiten! See the following pages for more information! Lieferung & Zahlungsart Lieferung: frei Haus Bestellung auf Rechnung SZABO-SCANDIC Lieferung: € 10,- HandelsgmbH & Co KG Erstbestellung Vorauskassa Quellenstraße 110, A-1100 Wien T. +43(0)1 489 3961-0 Zuschläge F. +43(0)1 489 3961-7 [email protected] • Mindermengenzuschlag www.szabo-scandic.com • Trockeneiszuschlag • Gefahrgutzuschlag linkedin.com/company/szaboscandic • Expressversand facebook.com/szaboscandic OR2G2 (Human) Recombinant Gene Summary: Olfactory receptors interact with Protein (P01) odorant molecules in the nose, to initiate a neuronal response that triggers the perception of a smell. The Catalog Number: H00081470-P01 olfactory receptor proteins are members of a large family of G-protein-coupled receptors (GPCR) arising from Regulation Status: For research use only (RUO) single coding-exon genes. Olfactory receptors share a 7-transmembrane domain structure with many Product Description: Human OR2G2 full-length ORF ( neurotransmitter and hormone receptors and are NP_001001915.1, 1 a.a. - 317 a.a.) recombinant protein responsible for the recognition and G protein-mediated with GST-tag at N-terminal. transduction of odorant signals. The olfactory receptor gene family is the largest in the genome. The Sequence: nomenclature assigned to the olfactory receptor genes MGMVRHTNESNLAGFILLGFSDYPQLQKVLFVLILILYL -

Cellular and Molecular Signatures in the Disease Tissue of Early
Cellular and Molecular Signatures in the Disease Tissue of Early Rheumatoid Arthritis Stratify Clinical Response to csDMARD-Therapy and Predict Radiographic Progression Frances Humby1,* Myles Lewis1,* Nandhini Ramamoorthi2, Jason Hackney3, Michael Barnes1, Michele Bombardieri1, Francesca Setiadi2, Stephen Kelly1, Fabiola Bene1, Maria di Cicco1, Sudeh Riahi1, Vidalba Rocher-Ros1, Nora Ng1, Ilias Lazorou1, Rebecca E. Hands1, Desiree van der Heijde4, Robert Landewé5, Annette van der Helm-van Mil4, Alberto Cauli6, Iain B. McInnes7, Christopher D. Buckley8, Ernest Choy9, Peter Taylor10, Michael J. Townsend2 & Costantino Pitzalis1 1Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. Departments of 2Biomarker Discovery OMNI, 3Bioinformatics and Computational Biology, Genentech Research and Early Development, South San Francisco, California 94080 USA 4Department of Rheumatology, Leiden University Medical Center, The Netherlands 5Department of Clinical Immunology & Rheumatology, Amsterdam Rheumatology & Immunology Center, Amsterdam, The Netherlands 6Rheumatology Unit, Department of Medical Sciences, Policlinico of the University of Cagliari, Cagliari, Italy 7Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow G12 8TA, UK 8Rheumatology Research Group, Institute of Inflammation and Ageing (IIA), University of Birmingham, Birmingham B15 2WB, UK 9Institute of -

NIH Public Access Author Manuscript Psychiatry Res
NIH Public Access Author Manuscript Psychiatry Res. Author manuscript; available in PMC 2014 November 30. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Psychiatry Res. 2013 November 30; 210(1): 287–293. doi:10.1016/j.psychres.2013.05.026. Assessment of gene expression in peripheral blood using RNAseq before and after weight restoration in anorexia nervosa Yunjung Kima,1, Sara Elizabeth Traceb,1, James Joseph Crowleya, Kimberly Ann Brownleyb, Robert Mark Hamerb,c, David Stephen Pisetskyd,e,f, Patrick Francis Sullivana,b, and Cynthia Marie Bulikb,g,* aDepartment of Genetics, University of North Carolina at Chapel Hill, NC, USA bDepartment of Psychiatry, University of North Carolina, Chapel Hill, NC, USA cDepartment of Biostatistics, University of North Carolina, Chapel Hill, NC, USA dDepartment of Medicine, Duke University, Durham, NC, USA eDepartment of Immunology, Duke University, Durham, NC, USA fMedical Research Service, Durham Veterans Administration Medical Center, Durham, NC, USA gDepartment of Nutrition, University of North Carolina, Chapel Hill, NC, USA Abstract We examined gene expression in the blood of six females with anorexia nervosa (AN) before and after weight restoration using RNAseq. AN cases (aged 19-39) completed clinical assessments and had blood drawn for RNA at hospital admission (T1, < ~75% ideal body weight, IBW) and again at discharge (T2, ≥ ~85% IBW). To examine the relationship between weight restoration and differential gene expression, normalized gene expression levels were analyzed using a paired design. We found 564 genes whose expression was nominally significantly different following weight restoration (p < 0.01, 231 increased and 333 decreased). -

Supplementary Table 1. Pain and PTSS Associated Genes (N = 604
Supplementary Table 1. Pain and PTSS associated genes (n = 604) compiled from three established pain gene databases (PainNetworks,[61] Algynomics,[52] and PainGenes[42]) and one PTSS gene database (PTSDgene[88]). These genes were used in in silico analyses aimed at identifying miRNA that are predicted to preferentially target this list genes vs. a random set of genes (of the same length). ABCC4 ACE2 ACHE ACPP ACSL1 ADAM11 ADAMTS5 ADCY5 ADCYAP1 ADCYAP1R1 ADM ADORA2A ADORA2B ADRA1A ADRA1B ADRA1D ADRA2A ADRA2C ADRB1 ADRB2 ADRB3 ADRBK1 ADRBK2 AGTR2 ALOX12 ANO1 ANO3 APOE APP AQP1 AQP4 ARL5B ARRB1 ARRB2 ASIC1 ASIC2 ATF1 ATF3 ATF6B ATP1A1 ATP1B3 ATP2B1 ATP6V1A ATP6V1B2 ATP6V1G2 AVPR1A AVPR2 BACE1 BAMBI BDKRB2 BDNF BHLHE22 BTG2 CA8 CACNA1A CACNA1B CACNA1C CACNA1E CACNA1G CACNA1H CACNA2D1 CACNA2D2 CACNA2D3 CACNB3 CACNG2 CALB1 CALCRL CALM2 CAMK2A CAMK2B CAMK4 CAT CCK CCKAR CCKBR CCL2 CCL3 CCL4 CCR1 CCR7 CD274 CD38 CD4 CD40 CDH11 CDK5 CDK5R1 CDKN1A CHRM1 CHRM2 CHRM3 CHRM5 CHRNA5 CHRNA7 CHRNB2 CHRNB4 CHUK CLCN6 CLOCK CNGA3 CNR1 COL11A2 COL9A1 COMT COQ10A CPN1 CPS1 CREB1 CRH CRHBP CRHR1 CRHR2 CRIP2 CRYAA CSF2 CSF2RB CSK CSMD1 CSNK1A1 CSNK1E CTSB CTSS CX3CL1 CXCL5 CXCR3 CXCR4 CYBB CYP19A1 CYP2D6 CYP3A4 DAB1 DAO DBH DBI DICER1 DISC1 DLG2 DLG4 DPCR1 DPP4 DRD1 DRD2 DRD3 DRD4 DRGX DTNBP1 DUSP6 ECE2 EDN1 EDNRA EDNRB EFNB1 EFNB2 EGF EGFR EGR1 EGR3 ENPP2 EPB41L2 EPHB1 EPHB2 EPHB3 EPHB4 EPHB6 EPHX2 ERBB2 ERBB4 EREG ESR1 ESR2 ETV1 EZR F2R F2RL1 F2RL2 FAAH FAM19A4 FGF2 FKBP5 FLOT1 FMR1 FOS FOSB FOSL2 FOXN1 FRMPD4 FSTL1 FYN GABARAPL1 GABBR1 GABBR2 GABRA2 GABRA4 -

Supplementary Methods
Heterogeneous Contribution of Microdeletions in the Development of Common Generalized and Focal epilepsies. SUPPLEMENTARY METHODS Epilepsy subtype extended description. Genetic Gereralized Epilepsy (GGE): Features unprovoked tonic and/or clonic seizures, originated inconsistently at some focal point within the brain that rapidly generalizes engaging bilateral distributed spikes and waves discharges on the electroencephalogram. This generalization can include cortical and sub cortical structures but not necessarily the entire cortex[1]. GGE is the most common group of epilepsies accounting for 20% of all cases[2]. It is characterized by an age-related onset and a strong familial aggregation and heritability which allows the assumption of a genetic cause. Although genetic associations have been identified, a broad spectrum of causes is acknowledged and remains largely unsolved [3]. Rolandic Epilepsy (RE): Commonly known also as Benign Epilepsy with Centrotemporal Spikes (BECTS), hallmarks early onset diagnosis (mean onset = 7 years old) with brief, focal hemifacial or oropharyngeal sensorimotor seizures alongside speech arrest and secondarily generalized tonic– clonic seizures, which mainly occur during sleep[4]. Rolandic epilepsy features a broad spectrum of less benign related syndromes called atypical Rolandic epilepsy (ARE), including benign partial epilepsy (ABPE), Landau–Kleffner syndrome(LKS) and epileptic encephalopathy with continuous spike-and-waves during sleep (CSWSS)[5]. Together they are the most common childhood epilepsy with a prevalence of 0.2–0.73/1000 (i.e. _1/2500)[6]. Adult Focal Epilepsy (AFE). Focal epilepsy is characterized by sporadic events of seizures originated within a specific brain region and restricted to one hemisphere. Although they can exhibit more than one network of wave discharges on the electroencephalogram, and different degrees of spreading, they feature a consistent site of origin. -

Identification of Key Genes and Pathways Involved in Response To
Deng et al. Biol Res (2018) 51:25 https://doi.org/10.1186/s40659-018-0174-7 Biological Research RESEARCH ARTICLE Open Access Identifcation of key genes and pathways involved in response to pain in goat and sheep by transcriptome sequencing Xiuling Deng1,2†, Dong Wang3†, Shenyuan Wang1, Haisheng Wang2 and Huanmin Zhou1* Abstract Purpose: This aim of this study was to investigate the key genes and pathways involved in the response to pain in goat and sheep by transcriptome sequencing. Methods: Chronic pain was induced with the injection of the complete Freund’s adjuvant (CFA) in sheep and goats. The animals were divided into four groups: CFA-treated sheep, control sheep, CFA-treated goat, and control goat groups (n 3 in each group). The dorsal root ganglions of these animals were isolated and used for the construction of a cDNA= library and transcriptome sequencing. Diferentially expressed genes (DEGs) were identifed in CFA-induced sheep and goats and gene ontology (GO) enrichment analysis was performed. Results: In total, 1748 and 2441 DEGs were identifed in CFA-treated goat and sheep, respectively. The DEGs identi- fed in CFA-treated goats, such as C-C motif chemokine ligand 27 (CCL27), glutamate receptor 2 (GRIA2), and sodium voltage-gated channel alpha subunit 3 (SCN3A), were mainly enriched in GO functions associated with N-methyl- D-aspartate (NMDA) receptor, infammatory response, and immune response. The DEGs identifed in CFA-treated sheep, such as gamma-aminobutyric acid (GABA)-related DEGs (gamma-aminobutyric acid type A receptor gamma 3 subunit [GABRG3], GABRB2, and GABRB1), SCN9A, and transient receptor potential cation channel subfamily V member 1 (TRPV1), were mainly enriched in GO functions related to neuroactive ligand-receptor interaction, NMDA receptor, and defense response. -

Olfactory Receptors in Non-Chemosensory Organs: the Nervous System in Health and Disease
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Sissa Digital Library REVIEW published: 05 July 2016 doi: 10.3389/fnagi.2016.00163 Olfactory Receptors in Non-Chemosensory Organs: The Nervous System in Health and Disease Isidro Ferrer 1,2,3*, Paula Garcia-Esparcia 1,2,3, Margarita Carmona 1,2,3, Eva Carro 2,4, Eleonora Aronica 5, Gabor G. Kovacs 6, Alice Grison 7 and Stefano Gustincich 7 1 Institute of Neuropathology, Bellvitge University Hospital, Hospitalet de Llobregat, University of Barcelona, Barcelona, Spain, 2 Center for Biomedical Research in Neurodegenerative Diseases (CIBERNED), Madrid, Spain, 3 Bellvitge Biomedical Research Institute (IDIBELL), Hospitalet de Llobregat, Barcelona, Spain, 4 Neuroscience Group, Research Institute Hospital, Madrid, Spain, 5 Department of Neuropathology, Academic Medical Center, University of Amsterdam, Amsterdam, Netherlands, 6 Institute of Neurology, Medical University of Vienna, Vienna, Austria, 7 Scuola Internazionale Superiore di Studi Avanzati (SISSA), Area of Neuroscience, Trieste, Italy Olfactory receptors (ORs) and down-stream functional signaling molecules adenylyl cyclase 3 (AC3), olfactory G protein a subunit (Gaolf), OR transporters receptor transporter proteins 1 and 2 (RTP1 and RTP2), receptor expression enhancing protein 1 (REEP1), and UDP-glucuronosyltransferases (UGTs) are expressed in neurons of the human and murine central nervous system (CNS). In vitro studies have shown that these receptors react to external stimuli and therefore are equipped to be functional. However, ORs are not directly related to the detection of odors. Several molecules delivered from the blood, cerebrospinal fluid, neighboring local neurons and glial cells, distant cells through the extracellular space, and the cells’ own self-regulating internal homeostasis Edited by: Filippo Tempia, can be postulated as possible ligands. -

Mouse Glra3 Conditional Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Glra3 Conditional Knockout Project (CRISPR/Cas9) Objective: To create a Glra3 conditional knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Glra3 gene (NCBI Reference Sequence: NM_080438 ; Ensembl: ENSMUSG00000038257 ) is located on Mouse chromosome 8. 10 exons are identified, with the ATG start codon in exon 1 and the TAA stop codon in exon 10 (Transcript: ENSMUST00000000275). Exon 3 will be selected as conditional knockout region (cKO region). Deletion of this region should result in the loss of function of the Mouse Glra3 gene. To engineer the targeting vector, homologous arms and cKO region will be generated by PCR using BAC clone RP24-365J12 as template. Cas9, gRNA and targeting vector will be co-injected into fertilized eggs for cKO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Homozygous null mice are fertile and display decreased inflammatory pain sensitization without any gross abnormalities in the brain or spinal cord. Exon 3 starts from about 17.22% of the coding region. The knockout of Exon 3 will result in frameshift of the gene. The size of intron 2 for 5'-loxP site insertion: 13863 bp, and the size of intron 3 for 3'-loxP site insertion: 34397 bp. The size of effective cKO region: ~568 bp. The cKO region does not have any other known gene. Page 1 of 7 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele gRNA region 5' gRNA region 3' 1 3 10 Targeting vector Targeted allele Constitutive KO allele (After Cre recombination) Legends Exon of mouse Glra3 Homology arm cKO region loxP site Page 2 of 7 https://www.alphaknockout.com Overview of the Dot Plot Window size: 10 bp Forward Reverse Complement Sequence 12 Note: The sequence of homologous arms and cKO region is aligned with itself to determine if there are tandem repeats. -
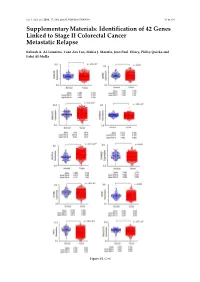
Identification of 42 Genes Linked to Stage II Colorectal Cancer Metastatic Relapse
Int. J. Mol. Sci. 2016, 17, 598; doi:10.3390/ijms17040598 S1 of S16 Supplementary Materials: Identification of 42 Genes Linked to Stage II Colorectal Cancer Metastatic Relapse Rabeah A. Al-Temaimi, Tuan Zea Tan, Makia J. Marafie, Jean Paul Thiery, Philip Quirke and Fahd Al-Mulla Figure S1. Cont. Int. J. Mol. Sci. 2016, 17, 598; doi:10.3390/ijms17040598 S2 of S16 Figure S1. Mean expression levels of fourteen genes of significant association with CRC DFS and OS that are differentially expressed in normal colon compared to CRC tissues. Each dot represents a sample. Table S1. Copy number aberrations associated with poor disease-free survival and metastasis in early stage II CRC as predicted by STAC and SPPS combined methodologies with resident gene symbols. CN stands for copy number, whereas CNV is copy number variation. Region Cytoband % of CNV Count of Region Event Gene Symbols Length Location Overlap Genes chr1:113,025,076–113,199,133 174,057 p13.2 CN Loss 0.0 2 AKR7A2P1, SLC16A1 chr1:141,465,960–141,822,265 356,305 q12–q21.1 CN Gain 95.9 1 SRGAP2B MIR5087, LOC10013000 0, FLJ39739, LOC10028679 3, PPIAL4G, PPIAL4A, NBPF14, chr1:144,911,564–146,242,907 1,331,343 q21.1 CN Gain 99.6 16 NBPF15, NBPF16, PPIAL4E, NBPF16, PPIAL4D, PPIAL4F, LOC645166, LOC388692, FCGR1C chr1:177,209,428–177,226,812 17,384 q25.3 CN Gain 0.0 0 chr1:197,652,888–197,676,831 23,943 q32.1 CN Gain 0.0 1 KIF21B chr1:201,015,278–201,033,308 18,030 q32.1 CN Gain 0.0 1 PLEKHA6 chr1:201,289,154–201,298,247 9093 q32.1 CN Gain 0.0 0 chr1:216,820,186–217,043,421 223,235 q41 CN -

Identification of Candidate Biomarkers and Pathways Associated with Type 1 Diabetes Mellitus Using Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2021.06.08.447531; this version posted June 9, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Identification of candidate biomarkers and pathways associated with type 1 diabetes mellitus using bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karnataka, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2021.06.08.447531; this version posted June 9, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Type 1 diabetes mellitus (T1DM) is a metabolic disorder for which the underlying molecular mechanisms remain largely unclear. This investigation aimed to elucidate essential candidate genes and pathways in T1DM by integrated bioinformatics analysis. In this study, differentially expressed genes (DEGs) were analyzed using DESeq2 of R package from GSE162689 of the Gene Expression Omnibus (GEO). Gene ontology (GO) enrichment analysis, REACTOME pathway enrichment analysis, and construction and analysis of protein-protein interaction (PPI) network, modules, miRNA-hub gene regulatory network and TF-hub gene regulatory network, and validation of hub genes were then performed. A total of 952 DEGs (477 up regulated and 475 down regulated genes) were identified in T1DM. GO and REACTOME enrichment result results showed that DEGs mainly enriched in multicellular organism development, detection of stimulus, diseases of signal transduction by growth factor receptors and second messengers, and olfactory signaling pathway.