RHO Binding to FAM65A Regulates Golgi Reorientation During Cell Migration Faraz K
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

MAP4K4 Expression in Cardiomyocytes: Multiple Isoforms, Multiple Phosphorylations and Interactions with Striatins
MAP4K4 expression in cardiomyocytes: multiple isoforms, multiple phosphorylations and interactions with striatins Article Published Version Creative Commons: Attribution 4.0 (CC-BY) Open access Fuller, S. J., Edmunds, N. S., McGuffin, L. J. ORCID: https://orcid.org/0000-0003-4501-4767, Hardyman, M. A., Cull, J. J., Alharbi, H. O., Meijles, D. N., Sugden, P. H. and Clerk, A. ORCID: https://orcid.org/0000-0002-5658-0708 (2021) MAP4K4 expression in cardiomyocytes: multiple isoforms, multiple phosphorylations and interactions with striatins. Biochemical Journal, 478 (11). pp. 2121-2143. ISSN 0264- 6021 doi: https://doi.org/10.1042/BCJ20210003 Available at http://centaur.reading.ac.uk/98442/ It is advisable to refer to the publisher’s version if you intend to cite from the work. See Guidance on citing . Published version at: http://dx.doi.org/10.1042/BCJ20210003 To link to this article DOI: http://dx.doi.org/10.1042/BCJ20210003 Publisher: Portland Press All outputs in CentAUR are protected by Intellectual Property Rights law, including copyright law. Copyright and IPR is retained by the creators or other copyright holders. Terms and conditions for use of this material are defined in the End User Agreement . www.reading.ac.uk/centaur CentAUR Central Archive at the University of Reading Reading’s research outputs online Biochemical Journal (2021) 478 2121–2143 https://doi.org/10.1042/BCJ20210003 Research Article MAP4K4 expression in cardiomyocytes: multiple isoforms, multiple phosphorylations and interactions with striatins Stephen J. Fuller1, Nick S. Edmunds1, Liam J. McGuffin1, Michelle A. Hardyman1, Joshua J. Cull1, Hajed O. Alharbi1, Daniel N. Meijles2, Peter H. -

Increased Expression of the Tail-Anchored Membrane Protein SLMAP in Adipose Tissue from Type 2 Tally Ho Diabetic Mice
Hindawi Publishing Corporation Experimental Diabetes Research Volume 2011, Article ID 421982, 10 pages doi:10.1155/2011/421982 Research Article Increased Expression of the Tail-Anchored Membrane Protein SLMAP in Adipose Tissue from Type 2 Tally Ho Diabetic Mice Xiaoliang Chen1 and Hong Ding2 1 Second People Hospital of Hangzhou, No. 1 Wenzhou Road, Hangzhou 310015, China 2 Department of Pharmacology, Weill Cornell Medical College in Qatar, P.O. Box 24144, Doha, Qatar Correspondence should be addressed to Hong Ding, [email protected] Received 6 April 2011; Accepted 5 May 2011 Academic Editor: N. Cameron Copyright © 2011 X. Chen and H. Ding. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. The tail-anchored membrane protein, sarcolemmal membrane associated protein (SLMAP) is encoded to a single gene that maps to the chromosome 3p14 region and has also been reported in certain diabetic populations. Our previous studies with db/db mice shown that a deregulation of SLMAP expression plays an important role in type 2 diabetes. Male Tally Ho mice were bred to present with either normoglycemia (NG) or hyperglycemia (HG). Abdominal adipose tissue from male Tally Ho mice of the HG group was found to have a significantly lower expression of the membrane associated glucose transporter-4 (GLUT-4) and higher expression of SLMAP compared to tissue from NG mice. There were 3 isoforms expressed in the abdominal adipose tissue, but only 45 kDa isoform of SLMAP was associated with the GLUT-4 revealed by immunoprecipitation data. -

Supplementary Table S1. Upregulated Genes Differentially
Supplementary Table S1. Upregulated genes differentially expressed in athletes (p < 0.05 and 1.3-fold change) Gene Symbol p Value Fold Change 221051_s_at NMRK2 0.01 2.38 236518_at CCDC183 0.00 2.05 218804_at ANO1 0.00 2.05 234675_x_at 0.01 2.02 207076_s_at ASS1 0.00 1.85 209135_at ASPH 0.02 1.81 228434_at BTNL9 0.03 1.81 229985_at BTNL9 0.01 1.79 215795_at MYH7B 0.01 1.78 217979_at TSPAN13 0.01 1.77 230992_at BTNL9 0.01 1.75 226884_at LRRN1 0.03 1.74 220039_s_at CDKAL1 0.01 1.73 236520_at 0.02 1.72 219895_at TMEM255A 0.04 1.72 201030_x_at LDHB 0.00 1.69 233824_at 0.00 1.69 232257_s_at 0.05 1.67 236359_at SCN4B 0.04 1.64 242868_at 0.00 1.63 1557286_at 0.01 1.63 202780_at OXCT1 0.01 1.63 1556542_a_at 0.04 1.63 209992_at PFKFB2 0.04 1.63 205247_at NOTCH4 0.01 1.62 1554182_at TRIM73///TRIM74 0.00 1.61 232892_at MIR1-1HG 0.02 1.61 204726_at CDH13 0.01 1.6 1561167_at 0.01 1.6 1565821_at 0.01 1.6 210169_at SEC14L5 0.01 1.6 236963_at 0.02 1.6 1552880_at SEC16B 0.02 1.6 235228_at CCDC85A 0.02 1.6 1568623_a_at SLC35E4 0.00 1.59 204844_at ENPEP 0.00 1.59 1552256_a_at SCARB1 0.02 1.59 1557283_a_at ZNF519 0.02 1.59 1557293_at LINC00969 0.03 1.59 231644_at 0.01 1.58 228115_at GAREM1 0.01 1.58 223687_s_at LY6K 0.02 1.58 231779_at IRAK2 0.03 1.58 243332_at LOC105379610 0.04 1.58 232118_at 0.01 1.57 203423_at RBP1 0.02 1.57 AMY1A///AMY1B///AMY1C///AMY2A///AMY2B// 208498_s_at 0.03 1.57 /AMYP1 237154_at LOC101930114 0.00 1.56 1559691_at 0.01 1.56 243481_at RHOJ 0.03 1.56 238834_at MYLK3 0.01 1.55 213438_at NFASC 0.02 1.55 242290_at TACC1 0.04 1.55 ANKRD20A1///ANKRD20A12P///ANKRD20A2/// -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

High-Throughput Discovery of Novel Developmental Phenotypes
High-throughput discovery of novel developmental phenotypes The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Dickinson, M. E., A. M. Flenniken, X. Ji, L. Teboul, M. D. Wong, J. K. White, T. F. Meehan, et al. 2016. “High-throughput discovery of novel developmental phenotypes.” Nature 537 (7621): 508-514. doi:10.1038/nature19356. http://dx.doi.org/10.1038/nature19356. Published Version doi:10.1038/nature19356 Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:32071918 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Nature. Manuscript Author Author manuscript; Manuscript Author available in PMC 2017 March 14. Published in final edited form as: Nature. 2016 September 22; 537(7621): 508–514. doi:10.1038/nature19356. High-throughput discovery of novel developmental phenotypes A full list of authors and affiliations appears at the end of the article. Abstract Approximately one third of all mammalian genes are essential for life. Phenotypes resulting from mouse knockouts of these genes have provided tremendous insight into gene function and congenital disorders. As part of the International Mouse Phenotyping Consortium effort to generate and phenotypically characterize 5000 knockout mouse lines, we have identified 410 Users may view, print, copy, and download text and data-mine the content in such documents, for the purposes of academic research, subject always to the full Conditions of use:http://www.nature.com/authors/editorial_policies/license.html#terms #Corresponding author: [email protected]. -

Detection of Aneuploidies by Paralogous Sequence Quantification S Deutsch, U Choudhury, G Merla, C Howald, a Sylvan, S E Antonarakis
908 J Med Genet: first published as 10.1136/jmg.2004.023184 on 9 December 2004. Downloaded from ORIGINAL ARTICLE Detection of aneuploidies by paralogous sequence quantification S Deutsch, U Choudhury, G Merla, C Howald, A Sylvan, S E Antonarakis ............................................................................................................................... J Med Genet 2004;41:908–915. doi: 10.1136/jmg.2004.023184 Background: Chromosomal aneuploidies are a common cause of congenital disorders associated with cognitive impairment and multiple dysmorphic features. Pre-natal diagnosis of aneuploidies is most See end of article for commonly performed by the karyotyping of fetal cells obtained by amniocentesis or chorionic villus authors’ affiliations sampling, but this method is labour intensive and requires about 14 days to complete. ....................... Methods: We have developed a PCR based method for the detection of targeted chromosome number Correspondence to: abnormalities termed paralogous sequence quantification (PSQ), based on the use of paralogous genes. Professor Stylianos E Paralogous sequences have a high degree of sequence identity, but accumulate nucleotide substitutions in Antonarakis, Department a locus specific manner. These sequence differences, which we term paralogous sequence mismatches of Genetic Medicine and Development, University of (PSMs), can be quantified using pyrosequencing technology, to estimate the relative dosage between Geneva Medical School, different chromosomes. We designed 10 assays for the detection of trisomies of chromosomes 13, 18, and GE 1211, Geneva, 21 and sex chromosome aneuploidies. Switzerland; Stylianos. antonarakis@medecine. Results: We evaluated the performance of this method on 175 DNAs, highly enriched for abnormal unige.ch samples. A correct and unambiguous diagnosis was given for 119 out of 120 aneuploid samples as well as for all the controls. -
![Downloaded from [266]](https://docslib.b-cdn.net/cover/7352/downloaded-from-266-347352.webp)
Downloaded from [266]
Patterns of DNA methylation on the human X chromosome and use in analyzing X-chromosome inactivation by Allison Marie Cotton B.Sc., The University of Guelph, 2005 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in The Faculty of Graduate Studies (Medical Genetics) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) January 2012 © Allison Marie Cotton, 2012 Abstract The process of X-chromosome inactivation achieves dosage compensation between mammalian males and females. In females one X chromosome is transcriptionally silenced through a variety of epigenetic modifications including DNA methylation. Most X-linked genes are subject to X-chromosome inactivation and only expressed from the active X chromosome. On the inactive X chromosome, the CpG island promoters of genes subject to X-chromosome inactivation are methylated in their promoter regions, while genes which escape from X- chromosome inactivation have unmethylated CpG island promoters on both the active and inactive X chromosomes. The first objective of this thesis was to determine if the DNA methylation of CpG island promoters could be used to accurately predict X chromosome inactivation status. The second objective was to use DNA methylation to predict X-chromosome inactivation status in a variety of tissues. A comparison of blood, muscle, kidney and neural tissues revealed tissue-specific X-chromosome inactivation, in which 12% of genes escaped from X-chromosome inactivation in some, but not all, tissues. X-linked DNA methylation analysis of placental tissues predicted four times higher escape from X-chromosome inactivation than in any other tissue. Despite the hypomethylation of repetitive elements on both the X chromosome and the autosomes, no changes were detected in the frequency or intensity of placental Cot-1 holes. -

Novel Mutation and Three Other Sequence Variants Segregating with Phenotype at Keratoconus 13Q32 Susceptibility Locus
European Journal of Human Genetics (2012) 20, 389–397 & 2012 Macmillan Publishers Limited All rights reserved 1018-4813/12 www.nature.com/ejhg ARTICLE Novel mutation and three other sequence variants segregating with phenotype at keratoconus 13q32 susceptibility locus Marta Czugala1,6, Justyna A Karolak1,6, Dorota M Nowak1, Piotr Polakowski2, Jose Pitarque3, Andrea Molinari3, Malgorzata Rydzanicz1, Bassem A Bejjani4, Beatrice YJT Yue5, Jacek P Szaflik2 and Marzena Gajecka*,1 Keratoconus (KTCN), a non-inflammatory corneal disorder characterized by stromal thinning, represents a major cause of corneal transplantations. Genetic and environmental factors have a role in the etiology of this complex disease. Previously reported linkage analysis revealed that chromosomal region 13q32 is likely to contain causative gene(s) for familial KTCN. Consequently, we have chosen eight positional candidate genes in this region: MBNL1, IPO5, FARP1, RNF113B, STK24, DOCK9, ZIC5 and ZIC2, and sequenced all of them in 51 individuals from Ecuadorian KTCN families and 105 matching controls. The mutation screening identified one mutation and three sequence variants showing 100% segregation under a dominant model with KTCN phenotype in one large Ecuadorian family. These substitutions were found in three different genes: c.2262A4C (p.Gln754His) and c.720+43A4GinDOCK9; c.2377-132A4CinIPO5 and c.1053+29G4CinSTK24. PolyPhen analyses predicted that c.2262A4C (Gln754His) is possibly damaging for the protein function and structure. Our results suggest that c.2262A4C (p.Gln754His) -

Genome-Wide DNA Methylation Analysis of KRAS Mutant Cell Lines Ben Yi Tew1,5, Joel K
www.nature.com/scientificreports OPEN Genome-wide DNA methylation analysis of KRAS mutant cell lines Ben Yi Tew1,5, Joel K. Durand2,5, Kirsten L. Bryant2, Tikvah K. Hayes2, Sen Peng3, Nhan L. Tran4, Gerald C. Gooden1, David N. Buckley1, Channing J. Der2, Albert S. Baldwin2 ✉ & Bodour Salhia1 ✉ Oncogenic RAS mutations are associated with DNA methylation changes that alter gene expression to drive cancer. Recent studies suggest that DNA methylation changes may be stochastic in nature, while other groups propose distinct signaling pathways responsible for aberrant methylation. Better understanding of DNA methylation events associated with oncogenic KRAS expression could enhance therapeutic approaches. Here we analyzed the basal CpG methylation of 11 KRAS-mutant and dependent pancreatic cancer cell lines and observed strikingly similar methylation patterns. KRAS knockdown resulted in unique methylation changes with limited overlap between each cell line. In KRAS-mutant Pa16C pancreatic cancer cells, while KRAS knockdown resulted in over 8,000 diferentially methylated (DM) CpGs, treatment with the ERK1/2-selective inhibitor SCH772984 showed less than 40 DM CpGs, suggesting that ERK is not a broadly active driver of KRAS-associated DNA methylation. KRAS G12V overexpression in an isogenic lung model reveals >50,600 DM CpGs compared to non-transformed controls. In lung and pancreatic cells, gene ontology analyses of DM promoters show an enrichment for genes involved in diferentiation and development. Taken all together, KRAS-mediated DNA methylation are stochastic and independent of canonical downstream efector signaling. These epigenetically altered genes associated with KRAS expression could represent potential therapeutic targets in KRAS-driven cancer. Activating KRAS mutations can be found in nearly 25 percent of all cancers1. -

The MAP4K4-STRIPAK Complex Promotes Growth and Tissue Invasion In
bioRxiv preprint doi: https://doi.org/10.1101/2021.05.07.442906; this version posted May 8, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 The MAP4K4-STRIPAK complex promotes growth and tissue invasion in 2 medulloblastoma 3 Jessica Migliavacca1, Buket Züllig1, Charles Capdeville1, Michael Grotzer2 and Martin Baumgartner1,* 4 1 Division of Oncology, Children’s Research Center, University Children’s Hospital Zürich, Zürich, 5 Switzerland 6 2 Division of Oncology, University Children’s Hospital Zürich, Zürich, Switzerland 7 8 *e-mail: [email protected] 9 10 Abstract 11 Proliferation and motility are mutually exclusive biological processes associated with cancer that depend 12 on precise control of upstream signaling pathways with overlapping functionalities. We find that STRN3 13 and STRN4 scaffold subunits of the STRIPAK complex interact with MAP4K4 for pathway regulation in 14 medulloblastoma. Disruption of the MAP4K4-STRIPAK complex impairs growth factor-induced 15 migration and tissue invasion and stalls YAP/TAZ target gene expression and oncogenic growth. The 16 migration promoting functions of the MAP4K4-STRIPAK complex involve the activation of novel PKCs 17 and the phosphorylation of the membrane targeting S157 residue of VASP through MAP4K4. The anti- 18 proliferative effect of complex disruption is associated with reduced YAP/TAZ target gene expression 19 and results in repressed tumor growth in the brain tissue. This dichotomous functionality of the STRIPAK 20 complex in migration and proliferation control acts through MAP4K4 regulation in tumor cells and 21 provides relevant mechanistic insights into novel tumorigenic functions of the STRIPAK complex in 22 medulloblastoma. -
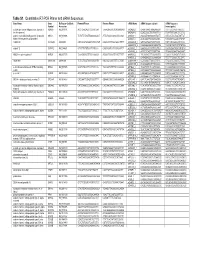
Table S1. Quantitative RT-PCR Primer and Sirna Sequences Gene Name
Table S1. Quantitative RT-PCR Primer and siRNA Sequences Gene Name Gene RefSeq or GenBank Forward Primer Reverse Primer siRNA Name siRNA Sequence (plus) siRNA Sequence Symbol Accession (minus/guide) a disintegrin and metalloproteinase domain 9 ADAM9 NM_003816 ACCTCAGCAGTTCCCATCAA TAAAGGAGGTGCAGGAGCAG siADAM9_1 GAGATTAACTAGAGAAAGA TCTTTCTCTAGTTAATCTC (meltrin gamma) siADAM9_2 GGAGGGAGTTCATAATTCA TGAATTATGAACTCCCTCC guanine nucleotide binding protein (G protein), GNAI3 NM_006496 TCTGTTATAGTTGGCGGCAGT ATTCTGCAAAGGCAAAAGGA siGNAI3_1 AGAACTAGCAGGAGTGATT AATCACTCCTGCTAGTTCT alpha inhibiting activity polypeptide 3 siGNAI3_2 ACACAGGTTCCAATACATA TATGTATTGGAACCTGTGT AA099748 AA099748 AA099748 CTACCACTGAGGTGTCCCTGT CAGCCATCTAACAGCATTTTT siAA099748_1 GTTAGATGGCTGATTAATA TATTAATCAGCCATCTAAC siAA099748_2 CGACAGAGGAGCAGCATTA TAATGCTGCTCCTCTGTCG calpain 12 CAPN12 NM_144691 GTCCTTCTGTCCCTCATCCA CAGCAGCTCCTCTGGAATCT siCAPN12_1 GGACACGCGTATTCCATCA TGATGGAATACGCGTGTCC siCAPN12_2 AATCCTCAGTTCCGTTTAA TTAAACGGAACTGAGGATT NMDA receptor regulated 1 NARG1 NM_057175 TAAAGGGAATTTGCCGAAGA AGCACTGGAGTTCGGTTTGT siNARG1_1 TGCGAGATCTTGAGGGTTA TAACCCTCAAGATCTCGCA siNARG1_2 GATAGGAGGTCCAAAAGAA TTCTTTTGGACCTCCTATC AK091308 AK091308 AK091308 TCTCCTGAATGGGAGGAATG GGCAACAGATGTTTCCTGTG siAK091308_1 CCAAAGACTTAGACTGTAA TTACAGTCTAAGTCTTTGG siAK091308_2 GCACAAAGCATTTACAACA TGTTGTAAATGCTTTGTGC serine/threonine kinase 24 (STE20 homolog, STK24 NM_003576 GCATCTGCCTTCCTTATCCA TGACAGTGTTTTGCCAGAGG siSTK24_1 TCGATTATCTCCATTCGGA TCCGAATGGAGATAATCGA yeast) siSTK24_2 CATCGGACTTGGACAGAAA -

Molecular Effects of Isoflavone Supplementation Human Intervention Studies and Quantitative Models for Risk Assessment
Molecular effects of isoflavone supplementation Human intervention studies and quantitative models for risk assessment Vera van der Velpen Thesis committee Promotors Prof. Dr Pieter van ‘t Veer Professor of Nutritional Epidemiology Wageningen University Prof. Dr Evert G. Schouten Emeritus Professor of Epidemiology and Prevention Wageningen University Co-promotors Dr Anouk Geelen Assistant professor, Division of Human Nutrition Wageningen University Dr Lydia A. Afman Assistant professor, Division of Human Nutrition Wageningen University Other members Prof. Dr Jaap Keijer, Wageningen University Dr Hubert P.J.M. Noteborn, Netherlands Food en Consumer Product Safety Authority Prof. Dr Yvonne T. van der Schouw, UMC Utrecht Dr Wendy L. Hall, King’s College London This research was conducted under the auspices of the Graduate School VLAG (Advanced studies in Food Technology, Agrobiotechnology, Nutrition and Health Sciences). Molecular effects of isoflavone supplementation Human intervention studies and quantitative models for risk assessment Vera van der Velpen Thesis submitted in fulfilment of the requirements for the degree of doctor at Wageningen University by the authority of the Rector Magnificus Prof. Dr M.J. Kropff, in the presence of the Thesis Committee appointed by the Academic Board to be defended in public on Friday 20 June 2014 at 13.30 p.m. in the Aula. Vera van der Velpen Molecular effects of isoflavone supplementation: Human intervention studies and quantitative models for risk assessment 154 pages PhD thesis, Wageningen University, Wageningen, NL (2014) With references, with summaries in Dutch and English ISBN: 978-94-6173-952-0 ABSTRact Background: Risk assessment can potentially be improved by closely linked experiments in the disciplines of epidemiology and toxicology.