Identification of Microrna-Related Tumorigenesis Variants and Genes in the Cancer Genome Atlas (TCGA) Data
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mechanical Forces Induce an Asthma Gene Signature in Healthy Airway Epithelial Cells Ayşe Kılıç1,10, Asher Ameli1,2,10, Jin-Ah Park3,10, Alvin T
www.nature.com/scientificreports OPEN Mechanical forces induce an asthma gene signature in healthy airway epithelial cells Ayşe Kılıç1,10, Asher Ameli1,2,10, Jin-Ah Park3,10, Alvin T. Kho4, Kelan Tantisira1, Marc Santolini 1,5, Feixiong Cheng6,7,8, Jennifer A. Mitchel3, Maureen McGill3, Michael J. O’Sullivan3, Margherita De Marzio1,3, Amitabh Sharma1, Scott H. Randell9, Jefrey M. Drazen3, Jefrey J. Fredberg3 & Scott T. Weiss1,3* Bronchospasm compresses the bronchial epithelium, and this compressive stress has been implicated in asthma pathogenesis. However, the molecular mechanisms by which this compressive stress alters pathways relevant to disease are not well understood. Using air-liquid interface cultures of primary human bronchial epithelial cells derived from non-asthmatic donors and asthmatic donors, we applied a compressive stress and then used a network approach to map resulting changes in the molecular interactome. In cells from non-asthmatic donors, compression by itself was sufcient to induce infammatory, late repair, and fbrotic pathways. Remarkably, this molecular profle of non-asthmatic cells after compression recapitulated the profle of asthmatic cells before compression. Together, these results show that even in the absence of any infammatory stimulus, mechanical compression alone is sufcient to induce an asthma-like molecular signature. Bronchial epithelial cells (BECs) form a physical barrier that protects pulmonary airways from inhaled irritants and invading pathogens1,2. Moreover, environmental stimuli such as allergens, pollutants and viruses can induce constriction of the airways3 and thereby expose the bronchial epithelium to compressive mechanical stress. In BECs, this compressive stress induces structural, biophysical, as well as molecular changes4,5, that interact with nearby mesenchyme6 to cause epithelial layer unjamming1, shedding of soluble factors, production of matrix proteins, and activation matrix modifying enzymes, which then act to coordinate infammatory and remodeling processes4,7–10. -

Environmental Influences on Endothelial Gene Expression
ENDOTHELIAL CELL GENE EXPRESSION John Matthew Jeff Herbert Supervisors: Prof. Roy Bicknell and Dr. Victoria Heath PhD thesis University of Birmingham August 2012 University of Birmingham Research Archive e-theses repository This unpublished thesis/dissertation is copyright of the author and/or third parties. The intellectual property rights of the author or third parties in respect of this work are as defined by The Copyright Designs and Patents Act 1988 or as modified by any successor legislation. Any use made of information contained in this thesis/dissertation must be in accordance with that legislation and must be properly acknowledged. Further distribution or reproduction in any format is prohibited without the permission of the copyright holder. ABSTRACT Tumour angiogenesis is a vital process in the pathology of tumour development and metastasis. Targeting markers of tumour endothelium provide a means of targeted destruction of a tumours oxygen and nutrient supply via destruction of tumour vasculature, which in turn ultimately leads to beneficial consequences to patients. Although current anti -angiogenic and vascular targeting strategies help patients, more potently in combination with chemo therapy, there is still a need for more tumour endothelial marker discoveries as current treatments have cardiovascular and other side effects. For the first time, the analyses of in-vivo biotinylation of an embryonic system is performed to obtain putative vascular targets. Also for the first time, deep sequencing is applied to freshly isolated tumour and normal endothelial cells from lung, colon and bladder tissues for the identification of pan-vascular-targets. Integration of the proteomic, deep sequencing, public cDNA libraries and microarrays, delivers 5,892 putative vascular targets to the science community. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Identification of Potential Markers for Type 2 Diabetes Mellitus Via Bioinformatics Analysis
1868 MOLECULAR MEDICINE REPORTS 22: 1868-1882, 2020 Identification of potential markers for type 2 diabetes mellitus via bioinformatics analysis YANA LU1, YIHANG LI1, GUANG LI1* and HAITAO LU2* 1Key Laboratory of Dai and Southern Medicine of Xishuangbanna Dai Autonomous Prefecture, Yunnan Branch, Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences and Peking Union Medical College, Jinghong, Yunnan 666100; 2Key Laboratory of Systems Biomedicine, Shanghai Center for Systems Biomedicine, Shanghai Jiao Tong University, Shanghai 200240, P.R. China Received March 20, 2019; Accepted January 20, 2020 DOI: 10.3892/mmr.2020.11281 Abstract. Type 2 diabetes mellitus (T2DM) is a multifactorial and cell proliferation’. These candidate genes were also involved in multigenetic disease, and its pathogenesis is complex and largely different signaling pathways associated with ‘PI3K/Akt signaling unknown. In the present study, microarray data (GSE201966) of pathway’, ‘Rap1 signaling pathway’, ‘Ras signaling pathway’ β-cell enriched tissue obtained by laser capture microdissection and ‘MAPK signaling pathway’, which are highly associated were downloaded, including 10 control and 10 type 2 diabetic with the development of T2DM. Furthermore, a microRNA subjects. A comprehensive bioinformatics analysis of microarray (miR)-target gene regulatory network and a transcription data in the context of protein-protein interaction (PPI) networks factor-target gene regulatory network were constructed based was employed, combined with subcellular location information on miRNet and NetworkAnalyst databases, respectively. to mine the potential candidate genes for T2DM and provide Notably, hsa-miR‑192-5p, hsa-miR‑124-5p and hsa-miR‑335-5p further insight on the possible mechanisms involved. First, appeared to be involved in T2DM by potentially regulating the differential analysis screened 108 differentially expressed expression of various candidate genes, including procollagen genes. -

Mutations in Mammalian Tolloid-Like 1 Gene Detected in Adult Patients with ASD
European Journal of Human Genetics (2009) 17, 344 – 351 & 2009 Macmillan Publishers Limited All rights reserved 1018-4813/09 $32.00 www.nature.com/ejhg ARTICLE Mutations in mammalian tolloid-like 1 gene detected in adult patients with ASD Paweł Stan´ czak1, Joanna Witecka2, Anna Szydło2, Ewa Gutmajster2, Małgorzata Lisik2, Aleksandra Augus´ciak-Duma2, Maciej Tarnowski2, Tomasz Czekaj2, Hanna Czekaj2 and Aleksander L Sieron´ *,2 1Swietokrzyskie Center for Cardiology, Regional Hospital, Kielce, Poland; 2Department of General and Molecular Biology and Genetics, CoE for Research and Teaching of Molecular Biology of Matrix and Nanotechnology, CoE Network, BioMedTech ‘Silesia’, Medical University of Silesia, Katowice, Poland Atrial septal defect (ASD) is an incomplete septation of atria in human heart causing circulatory problems. Its frequency is estimated at one per 10 000. Actions of numerous genes have been linked to heart development. However, no single gene defect causing ASD has yet been identified. Incomplete heart septation similar to ASD was reported in transgenic mice with both inactive alleles of gene encoding mammalian zinc metalloprotease a mammalian tolloid-like 1 (tll1). Here, we have screened 19 ASD patients and 15 healthy age-matched individuals for mutations in TLL1 gene. All 22 exons were analyzed exon by exon for heteroduplex formation. Subsequently, DNA fragments forming heteroduplexes were sequenced. In four nonrelated patients, three missense mutations in coding sequence, and one single base change in the 50UTR have been detected. Two mutations (Met182Leu, and Ala238Val) were detected in ASD patients with the same clinical phenotype. As the second mutation locates immediately upstream of the catalytic zinc-binding signature, it might change the enzyme substrate specificity. -
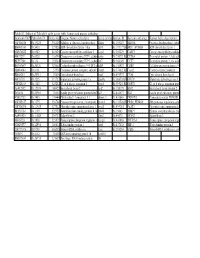
Table S3: Subset of Zebrafish Early Genes with Human And
Table S3: Subset of Zebrafish early genes with human and mouse orthologs Genbank ID(ZFZebrafish ID Entrez GenUnigene Name (zebrafish) Gene symbo Human ID Humann ortholog Human Gene description AW116838 Dr.19225 336425 Aldolase a, fructose-bisphosphate aldoa Hs.155247 ALDOA Fructose-bisphosphate aldola BM005100 Dr.5438 327026 ADP-ribosylation factor 1 like arf1l Hs.119177||HsARF1_HUMAN ADP-ribosylation factor 1 AW076882 Dr.6582 403025 Cancer susceptibility candidate 3 casc3 Hs.350229 CASC3 Cancer susceptibility candidat AI437239 Dr.6928 116994 Chaperonin containing TCP1, subun cct6a Hs.73072||Hs.CCT6A T-complex protein 1, zeta sub BE557308 Dr.134 192324 Chaperonin containing TCP1, subun cct7 Hs.368149 CCT7 T-complex protein 1, eta subu BG303647 Dr.26326 321602 Cyclin-dependent kinase 9 (CDC2-recdk9 Hs.150423 CDK9 Cell division protein kinase 9 AB040044 Dr.8161 57970 Coatomer protein complex, subunit zcopz1 Hs.37482||Hs.Copz2 Coatomer zeta-2 subunit BI888253 Dr.20911 30436 Eyes absent homolog 1 eya1 Hs.491997 EYA4 Eyes absent homolog 4 AI878758 Dr.3225 317737 Glutamate dehydrogenase 1a glud1a Hs.368538||HsGLUD1 Glutamate dehydrogenase 1, AW128619 Dr.1388 325284 G1 to S phase transition 1 gspt1 Hs.59523||Hs.GSPT1 G1 to S phase transition prote AF412832 Dr.12595 140427 Heat shock factor 2 hsf2 Hs.158195 HSF2 Heat shock factor protein 2 D38454 Dr.20916 30151 Insulin gene enhancer protein Islet3 isl3 Hs.444677 ISL2 Insulin gene enhancer protein AY052752 Dr.7485 170444 Pbx/knotted 1 homeobox 1.1 pknox1.1 Hs.431043 PKNOX1 Homeobox protein PKNOX1 -

Supplemental Table 1. Complete Gene Lists and GO Terms from Figure 3C
Supplemental Table 1. Complete gene lists and GO terms from Figure 3C. Path 1 Genes: RP11-34P13.15, RP4-758J18.10, VWA1, CHD5, AZIN2, FOXO6, RP11-403I13.8, ARHGAP30, RGS4, LRRN2, RASSF5, SERTAD4, GJC2, RHOU, REEP1, FOXI3, SH3RF3, COL4A4, ZDHHC23, FGFR3, PPP2R2C, CTD-2031P19.4, RNF182, GRM4, PRR15, DGKI, CHMP4C, CALB1, SPAG1, KLF4, ENG, RET, GDF10, ADAMTS14, SPOCK2, MBL1P, ADAM8, LRP4-AS1, CARNS1, DGAT2, CRYAB, AP000783.1, OPCML, PLEKHG6, GDF3, EMP1, RASSF9, FAM101A, STON2, GREM1, ACTC1, CORO2B, FURIN, WFIKKN1, BAIAP3, TMC5, HS3ST4, ZFHX3, NLRP1, RASD1, CACNG4, EMILIN2, L3MBTL4, KLHL14, HMSD, RP11-849I19.1, SALL3, GADD45B, KANK3, CTC- 526N19.1, ZNF888, MMP9, BMP7, PIK3IP1, MCHR1, SYTL5, CAMK2N1, PINK1, ID3, PTPRU, MANEAL, MCOLN3, LRRC8C, NTNG1, KCNC4, RP11, 430C7.5, C1orf95, ID2-AS1, ID2, GDF7, KCNG3, RGPD8, PSD4, CCDC74B, BMPR2, KAT2B, LINC00693, ZNF654, FILIP1L, SH3TC1, CPEB2, NPFFR2, TRPC3, RP11-752L20.3, FAM198B, TLL1, CDH9, PDZD2, CHSY3, GALNT10, FOXQ1, ATXN1, ID4, COL11A2, CNR1, GTF2IP4, FZD1, PAX5, RP11-35N6.1, UNC5B, NKX1-2, FAM196A, EBF3, PRRG4, LRP4, SYT7, PLBD1, GRASP, ALX1, HIP1R, LPAR6, SLITRK6, C16orf89, RP11-491F9.1, MMP2, B3GNT9, NXPH3, TNRC6C-AS1, LDLRAD4, NOL4, SMAD7, HCN2, PDE4A, KANK2, SAMD1, EXOC3L2, IL11, EMILIN3, KCNB1, DOK5, EEF1A2, A4GALT, ADGRG2, ELF4, ABCD1 Term Count % PValue Genes regulation of pathway-restricted GDF3, SMAD7, GDF7, BMPR2, GDF10, GREM1, BMP7, LDLRAD4, SMAD protein phosphorylation 9 6.34 1.31E-08 ENG pathway-restricted SMAD protein GDF3, SMAD7, GDF7, BMPR2, GDF10, GREM1, BMP7, LDLRAD4, phosphorylation -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig. -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -

Identification of Candidate Parkinson Disease Genes by Integrating Genome-Wide Association Study, Expression, and Epigenetic Data Sets
Research JAMA Neurology | Original Investigation Identification of Candidate Parkinson Disease Genes by Integrating Genome-Wide Association Study, Expression, and Epigenetic Data Sets Demis A. Kia, MBBS; David Zhang, MSc; Sebastian Guelfi, PhD; Claudia Manzoni, PhD; Leon Hubbard, PhD; Regina H. Reynolds, MSc; Juan Botía, PhD; Mina Ryten, MD; Raffaele Ferrari, PhD; Patrick A. Lewis, PhD; Nigel Williams, PhD; Daniah Trabzuni, PhD; John Hardy, PhD; Nicholas W. Wood, PhD; for the United Kingdom Brain Expression Consortium (UKBEC) and the International Parkinson’s Disease Genomics Consortium (IPDGC) Supplemental content IMPORTANCE Substantial genome-wide association study (GWAS) work in Parkinson disease (PD) has led to the discovery of an increasing number of loci shown reliably to be associated with increased risk of disease. Improved understanding of the underlying genes and mechanisms at these loci will be key to understanding the pathogenesis of PD. OBJECTIVE To investigate what genes and genomic processes underlie the risk of sporadic PD. DESIGN AND SETTING This genetic association study used the bioinformatic tools Coloc and transcriptome-wide association study (TWAS) to integrate PD case-control GWAS data published in 2017 with expression data (from Braineac, the Genotype-Tissue Expression [GTEx], and CommonMind) and methylation data (derived from UK Parkinson brain samples) to uncover putative gene expression and splicing mechanisms associated with PD GWAS signals. Candidate genes were further characterized using cell-type specificity, weighted gene coexpression networks, and weighted protein-protein interaction networks. MAIN OUTCOMES AND MEASURES It was hypothesized a priori that some genes underlying PD loci would alter PD risk through changes to expression, splicing, or methylation. -

GIPC2 Antibody
Product Datasheet GIPC2 Antibody Catalog No: #32240 Orders: [email protected] Description Support: [email protected] Product Name GIPC2 Antibody Host Species Rabbit Clonality Polyclonal Purification Antibodies were purified by affinity purification using immunogen. Applications WB IHC Species Reactivity Hu Ms Rt Specificity The antibody detects endogenous level of total GIPC2 protein. Immunogen Type Recombinant Protein Immunogen Description Recombinant protein of human GIPC2. Target Name GIPC2 Other Names GIPC2; FLJ20075; SEMCAP-2; SEMCAP2; Accession No. Swiss-Prot:Q8TF65NCBI Gene ID:54810 SDS-PAGE MW 34KD Concentration 1.0mg/ml Formulation Supplied at 1.0mg/mL in phosphate buffered saline (without Mg2+ and Ca2+), pH 7.4, 150mM NaCl, 0.02% sodium azide and 50% glycerol. Storage Store at -20°C Application Details Western blotting: 1:500 - 1:2000 Immunohistochemistry: 1:50 - 1:100 Images Western blot analysis of extracts of various cell lines, using GIPC2 antibody. Background The eukaryotic PDZ domain is a multifunctional protein-protein interacting motif that is found in a variety of proteins and is involved in both the clustering of signaling molecules and the organization of protein networks. GIPC2 (GIPC PDZ domain containing family, member 2), also known as Address: 8400 Baltimore Ave., Suite 302, College Park, MD 20740, USA http://www.sabbiotech.com 1 SEMCAP2, is a 315 amino acid protein that localizes to the cytoplasm and contains one PDZ domain. Expressed at high levels in kidney and colon and at lower levels in adult liver, GIPC2 interacts with SEMA5A and is thought to function as a scaffold protein, possibly modulating cell adhesion and growth factor signaling and playing a role in tumorigenesis. -

CCT6A Sirna (Mouse)
For research purposes only, not for human use Product Data Sheet CCT6A siRNA (Mouse) Catalog # Source Reactivity Applications CRM0566 Synthetic M RNAi Description siRNA to inhibit CCT6A expression using RNA interference Specificity CCT6A siRNA (Mouse) is a target-specific 19-23 nt siRNA oligo duplexes designed to knock down gene expression. Form Lyophilized powder Gene Symbol CCT6A Alternative Names CCT6; CCTZ; CCTZ1; T-complex protein 1 subunit zeta; TCP-1-zeta; CCT-zeta-1 Entrez Gene 12466 (Mouse) SwissProt P80317 (Mouse) Purity > 97% Quality Control Oligonucleotide synthesis is monitored base by base through trityl analysis to ensure appropriate coupling efficiency. The oligo is subsequently purified by affinity-solid phase extraction. The annealed RNA duplex is further analyzed by mass spectrometry to verify the exact composition of the duplex. Each lot is compared to the previous lot by mass spectrometry to ensure maximum lot-to-lot consistency. Components We offers pre-designed sets of 3 different target-specific siRNA oligo duplexes of mouse CCT6A gene. Each vial contains 5 nmol of lyophilized siRNA. The duplexes can be transfected individually or pooled together to achieve knockdown of the target gene, which is most commonly assessed by qPCR or western blot. Our siRNA oligos are also chemically modified (2’-OMe) at no extra charge for increased stability and enhanced knockdown in vitro and in vivo. Directions for Use We recommends transfection with 100 nM siRNA 48 to 72 hours prior to cell lysis. Application key: E- ELISA, WB-