WHO Guidelines for the Production, Control and Regulation of Snake Antivenom Immunoglobulins
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Redacted for Privacy
AN ABSTRACT OF THE THESIS OF Ian B. Edwards for the degree of Master of Arts in Applied Anthropology presented on April 30. 2003. Title: The Fetish Market and Animal Parts Trade of Mali. West Africa: An Ethnographic Investigation into Cultural Use and Significance. Abstract approved: Redacted for Privacy David While much research has examined the intricate interactions associated with the harvesting of wild animals for human consumption, little work has been undertaken in attempting to understand the greater socio-cultural significance of such use. In addition, to properly understand such systems of interaction, an intimate knowledge is required with regard to the rationale or motivation of resource users. In present day Mali, West Africa, the population perceives and upholds wildlife as a resource not only of valuable animal protein, in a region of famine and drought, but a means of generating income. The animal parts trade is but one mechanism within the larger socio-cultural structure that exploits wildlife through a complex human-environmental system to the benefit of those who participate. Moreover, this informal, yet highly structured system serves both cultural and outsider demand through its goods and services. By using traditional ethnographic investigation techniques (participant observation and semi-structured interviews) in combination with thick narration and multidisciplinary analysis (socio- cultural and biological-environmental), it is possible to construct a better understanding of the functions, processes, and motivation of those who participate. In a world where there is butonlya limited supply of natural and wild resources, understanding human- environmental systems is of critical value. ©Copyright by Ian B. -
Snake Bite Protocol
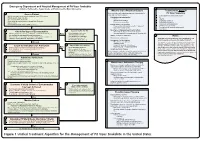
Lavonas et al. BMC Emergency Medicine 2011, 11:2 Page 4 of 15 http://www.biomedcentral.com/1471-227X/11/2 and other Rocky Mountain Poison and Drug Center treatment of patients bitten by coral snakes (family Ela- staff. The antivenom manufacturer provided funding pidae), nor by snakes that are not indigenous to the US. support. Sponsor representatives were not present dur- At the time this algorithm was developed, the only ing the webinar or panel discussions. Sponsor represen- antivenom commercially available for the treatment of tatives reviewed the final manuscript before publication pit viper envenomation in the US is Crotalidae Polyva- ® for the sole purpose of identifying proprietary informa- lent Immune Fab (ovine) (CroFab , Protherics, Nash- tion. No modifications of the manuscript were requested ville, TN). All treatment recommendations and dosing by the manufacturer. apply to this antivenom. This algorithm does not con- sider treatment with whole IgG antivenom (Antivenin Results (Crotalidae) Polyvalent, equine origin (Wyeth-Ayerst, Final unified treatment algorithm Marietta, Pennsylvania, USA)), because production of The unified treatment algorithm is shown in Figure 1. that antivenom has been discontinued and all extant The final version was endorsed unanimously. Specific lots have expired. This antivenom also does not consider considerations endorsed by the panelists are as follows: treatment with other antivenom products under devel- opment. Because the panel members are all hospital- Role of the unified treatment algorithm -

Mms NASICORNIS John En Marion Bakker, Con Reach up to 150 Cm in Length
mms NASICORNIS John en Marion Bakker, con reach up to 150 cm in length. They ore Spuistraat 119, regarded as the most beautifully coloured and 2987 TH Ridderkerk. marked vipers, but some specimens of the East The Netherlands. African Gaboon viper (Bilis gabonica gabonica) Phone: +31-180 413492. could very well compete for the title 'most E-mail: [email protected] beautiful viper of the world'. Recently we found on the Internet a number of The colours provide excellent camouflage on articles that could be used freely. We translated the forest floor where the snakes occur but in a the articles and added information and our well decorated cage you might also only find own experiences. them after taking a close look (but not to close, please). In their natural habitat they are very REGION OF ORIGIN AND HABITAT often covered by mud. Of course the colours will not be so very bright when this is the case. This big viper occurs in Central Africa, from Kenya in the east into Guinea in the west, and from THE POISON Sudan in the north to Angolo in the south. They live in tropical forests, especially in the vicinity of There is little knowledge about the poison of rivers, lakes, swamps and other damp places. Bilis nasicornis, although there are documen Bitis nosicornis con be regarded as semi-aquatic. ted bites. Generally the poison should be about the some as that of Bilis gabonica but, when SIZE AND APPEARANCE delivering a full bite, the dose of poison is often smaller. It is strongly haemotoxic so a An adult animal reaches a breathtaking size bite must always be treated as potentially let for a member of the viper family but is still hal, even with very young snakes. -

Molecular Characterization of Three Novel Phospholipase A2 Proteins from the Venom of Atheris Chlorechis, Atheris Nitschei and Atheris Squamigera
toxins Article Molecular Characterization of Three Novel Phospholipase A2 Proteins from the Venom of Atheris chlorechis, Atheris nitschei and Atheris squamigera He Wang 1,*, Xiaole Chen 2,*, Mei Zhou 3, Lei Wang 3, Tianbao Chen 3 and Chris Shaw 3 1 School of Integrative Medicine, Fujian University of Traditional Chinese Medicine, No.1 Qiu Yang Road, Shangjie Town, Fuzhou 350122, Fujian, China 2 School of Pharmacy, Fujian Medical University, No.1 Xueyuan Road, Shangjie Town, Fuzhou 350004, Fujian, China 3 Natural Drug Discovery Group, School of Pharmacy, Queen’s University Belfast, University Road, Belfast BT7 1NN, UK; [email protected] (M.Z.); [email protected] (L.W.); [email protected] (T.C.); [email protected] (C.S.) * Correspondence: [email protected] or [email protected] (H.W.); [email protected] (X.C.); Tel.: +86-591-2286-1151 (H.W.); +86-591-2286-2692 (X.C.) Academic Editor: Bryan Grieg Fry Received: 24 February 2016; Accepted: 20 May 2016; Published: 1 June 2016 Abstract: Secretory phospholipase A2 (sPLA2) is known as a major component of snake venoms and displays higher-order catalytic hydrolysis functions as well as a wide range of pathological effects. Atheris is not a notoriously dangerous genus of snakes although there are some reports of fatal cases after envenomation due to the effects of coagulation disturbances and hemorrhaging. Molecular characterization of Atheris venom enzymes is incomplete and there are only a few reports in the literature. Here, we report, for the first time, the cloning and characterization of three novel cDNAs encoding phospholipase A2 precursors (one each) from the venoms of the Western bush viper (Atheris chlorechis), the Great Lakes bush viper (Atheris nitschei) and the Variable bush viper (Atheris squamigera), using a “shotgun cloning” strategy. -

Broad-Headed Snake (Hoplocephalus Bungaroides)', Proceedings of the Royal Zoological Society of New South Wales (1946-7), Pp
Husbandry Guidelines Broad-Headed Snake Hoplocephalus bungaroides Compiler – Charles Morris Western Sydney Institute of TAFE, Richmond Captive Animals Certificate III RUV3020R Lecturers: Graeme Phipps, Jacki Salkeld & Brad Walker 2009 1 Occupational Health and Safety WARNING This Snake is DANGEROUSLY VENOMOUS CAPABLE OF INFLICTING A POTENTIALLY FATAL BITE ALWAYS HAVE A COMPRESSION BANDAGE WITHIN REACH SNAKE BITE TREATMENT: Do NOT wash the wound. Do NOT cut the wound, apply substances to the wound or use a tourniquet. Do NOT remove jeans or shirt as any movement will assist the venom to enter the blood stream. KEEP THE VICTIM STILL. 1. Apply a broad pressure bandage over the bite site as soon as possible. 2. Keep the limb still. The bandage should be as tight as you would bind a sprained ankle. 3. Extend the bandage down to the fingers or toes then up the leg as high as possible. (For a bite on the hand or forearm bind up to the elbow). 4. Apply a splint if possible, to immobilise the limb. 5. Bind it firmly to as much of the limb as possible. (Use a sling for an arm injury). Bring transport to the victim where possible or carry them to transportation. Transport the victim to the nearest hospital. Please Print this page off and put it up on the wall in your snake room. 2 There is some serious occupational health risks involved in keeping venomous snakes. All risk can be eliminated if kept clean and in the correct lockable enclosures with only the risk of handling left in play. -

Dwarfs on the Move: Spatial Ecology of the World's Smallest Viper, Bitis Schneideri Author(S) :Bryan Maritz and Graham J
Dwarfs on the Move: Spatial Ecology of the World's Smallest Viper, Bitis schneideri Author(s) :Bryan Maritz and Graham J. Alexander Source: Copeia, 2012(1):115-120. 2012. Published By: The American Society of Ichthyologists and Herpetologists DOI: http://dx.doi.org/10.1643/CH-11-048 URL: http://www.bioone.org/doi/full/10.1643/CH-11-048 BioOne (www.bioone.org) is a nonprofit, online aggregation of core research in the biological, ecological, and environmental sciences. BioOne provides a sustainable online platform for over 170 journals and books published by nonprofit societies, associations, museums, institutions, and presses. Your use of this PDF, the BioOne Web site, and all posted and associated content indicates your acceptance of BioOne’s Terms of Use, available at www.bioone.org/page/terms_of_use. Usage of BioOne content is strictly limited to personal, educational, and non-commercial use. Commercial inquiries or rights and permissions requests should be directed to the individual publisher as copyright holder. BioOne sees sustainable scholarly publishing as an inherently collaborative enterprise connecting authors, nonprofit publishers, academic institutions, research libraries, and research funders in the common goal of maximizing access to critical research. Copeia 2012, No. 1, 115–120 Dwarfs on the Move: Spatial Ecology of the World’s Smallest Viper, Bitis schneideri Bryan Maritz1 and Graham J. Alexander1 Namaqua Dwarf Adders (Bitis schneideri) are small viperids that inhabit sandy coastal habitats within the Succulent Karoo Biome in southern Africa. Their ecology, and the faunal ecology within the region in general, is poorly documented, hampering effective conservation planning for this biodiversity hotspot. -

An in Vivo Examination of the Differences Between Rapid
www.nature.com/scientificreports OPEN An in vivo examination of the diferences between rapid cardiovascular collapse and prolonged hypotension induced by snake venom Rahini Kakumanu1, Barbara K. Kemp-Harper1, Anjana Silva 1,2, Sanjaya Kuruppu3, Geofrey K. Isbister 1,4 & Wayne C. Hodgson1* We investigated the cardiovascular efects of venoms from seven medically important species of snakes: Australian Eastern Brown snake (Pseudonaja textilis), Sri Lankan Russell’s viper (Daboia russelii), Javanese Russell’s viper (D. siamensis), Gaboon viper (Bitis gabonica), Uracoan rattlesnake (Crotalus vegrandis), Carpet viper (Echis ocellatus) and Puf adder (Bitis arietans), and identifed two distinct patterns of efects: i.e. rapid cardiovascular collapse and prolonged hypotension. P. textilis (5 µg/kg, i.v.) and E. ocellatus (50 µg/kg, i.v.) venoms induced rapid (i.e. within 2 min) cardiovascular collapse in anaesthetised rats. P. textilis (20 mg/kg, i.m.) caused collapse within 10 min. D. russelii (100 µg/kg, i.v.) and D. siamensis (100 µg/kg, i.v.) venoms caused ‘prolonged hypotension’, characterised by a persistent decrease in blood pressure with recovery. D. russelii venom (50 mg/kg and 100 mg/kg, i.m.) also caused prolonged hypotension. A priming dose of P. textilis venom (2 µg/kg, i.v.) prevented collapse by E. ocellatus venom (50 µg/kg, i.v.), but had no signifcant efect on subsequent addition of D. russelii venom (1 mg/kg, i.v). Two priming doses (1 µg/kg, i.v.) of E. ocellatus venom prevented collapse by E. ocellatus venom (50 µg/kg, i.v.). B. gabonica, C. vegrandis and B. -

Xenosaurus Tzacualtipantecus. the Zacualtipán Knob-Scaled Lizard Is Endemic to the Sierra Madre Oriental of Eastern Mexico
Xenosaurus tzacualtipantecus. The Zacualtipán knob-scaled lizard is endemic to the Sierra Madre Oriental of eastern Mexico. This medium-large lizard (female holotype measures 188 mm in total length) is known only from the vicinity of the type locality in eastern Hidalgo, at an elevation of 1,900 m in pine-oak forest, and a nearby locality at 2,000 m in northern Veracruz (Woolrich- Piña and Smith 2012). Xenosaurus tzacualtipantecus is thought to belong to the northern clade of the genus, which also contains X. newmanorum and X. platyceps (Bhullar 2011). As with its congeners, X. tzacualtipantecus is an inhabitant of crevices in limestone rocks. This species consumes beetles and lepidopteran larvae and gives birth to living young. The habitat of this lizard in the vicinity of the type locality is being deforested, and people in nearby towns have created an open garbage dump in this area. We determined its EVS as 17, in the middle of the high vulnerability category (see text for explanation), and its status by the IUCN and SEMAR- NAT presently are undetermined. This newly described endemic species is one of nine known species in the monogeneric family Xenosauridae, which is endemic to northern Mesoamerica (Mexico from Tamaulipas to Chiapas and into the montane portions of Alta Verapaz, Guatemala). All but one of these nine species is endemic to Mexico. Photo by Christian Berriozabal-Islas. Amphib. Reptile Conserv. | http://redlist-ARC.org 01 June 2013 | Volume 7 | Number 1 | e61 Copyright: © 2013 Wilson et al. This is an open-access article distributed under the terms of the Creative Com- mons Attribution–NonCommercial–NoDerivs 3.0 Unported License, which permits unrestricted use for non-com- Amphibian & Reptile Conservation 7(1): 1–47. -

Sudan Snakes
NOTES ON SUDAN SNAKES A GUIDE TO THE SPECIES REPRESENTED IN THE COLLECTION IN THE NATURAL HISTORY MUSEUM KHARTOUM BY N. L. CORKILL, M.D. Sudan Medical Service. SUDAN GOVERNMENT MUSEUM (NATURAL HISTORY) PUBLICATION No. 3 AUGUST, 1935 PRICE - - - P:T. 10(2/-). NOTES ON SUDAN SNAKES A GUIDE TO THE SPECIES REPRESENTED IN THE COLLECTION IN THE NATURAL HISTORY MUSEUM KHARTOUM BY N. L. GORKILL, M.D. Sudan Medical Service. SUDAN GOVERNMENT MUSEUM (NATURAL HISTORY) PUBLICATION No. 3 AUGUST, 1935 PRICE - - - P.T. 10(2/-). PRINTED BY M*CORQUODALE & CO. J CONTENTS. PAGE Foreword ... ... ... ... ... ... ••• ••• 4 Preface ... 5 Introduction... ... ... ... ... ... ... ... 6 Systematic Index ... ... ... ... ... ... ... 9 Family Typhlopidge, the Blind Snakes 11 Family Leptotyphlopidse, the Earth Snakes ... ... ... n Family Boidae, the Boas and Pythons ... ... ... ... 12 Family Colubridae, the Colubrids 14 (a) Series Aglypha, the Fangless Colubrids ... ... 14 (b) Series Opisthoglypha, the Back-fanged Colubrids ... 19 Family Elapidae, the Cobras 23 Family Viperidas, the Vipers ... ... ... ... ... 26 Key to the Identification of Sudanese Thanat ophidians ... 31 Snake Bite in the Sudan 33 First Aid and Treatment of Snake Bite 34 Index to Snake Names :—• (a) Scientific ... ... ... ... ... ... ... 36 (b) Popular English 37 (c) Vernacular Sudanese 38 Appendix I. List of additional Sudanese Species ... ... 39 Appendix II. Instructions for Collectors 40 3 FOREWORD. HE Sudan Government collection of snakes was first started in 1920, and by 1930 contained 220 specimens. In the latter year T Mr. N. L. Corkill of the Sudan Medical Service, who had recently completed a survey of snakes and snake bite in Iraq, undertook to classify all specimens in the collection and subsequent additions thereto. -

Gunther the Tropical Moccasin
38 I Litteratura Serpentium, 1993, Vol. 13, Nr. 2 AGKISTRODON BILINEATUS - GUNTHER THE TROPICAL MOCCASIN By: Peet Smetsers, Langvennen Zuid 18, 5061 NR Oisterwijk, The Netherlands. Contents: Introduction - The species - Distribution and habitat - Venom toxicity and behaviour - Own experiences - References. * * * INTRODUCTION For over a year I have been in possession of a male and a female tropical moccasin (Agkistrodon bilineatus). During this period these snakes have become, next to my horned vipers (Cerastes cerastes karlhartli), my favourite animals. Therefore I offer the following for further consideration. THE SPECIES Agkistrodon bilineatus bilineatus belongs to the family of pitvipers, the Crotalidae. They are in possession of a heat sensitive organ both openings of which are found just under the eyes. Apart from Agkistrodon bilineatus bilineatus there are three other subspecies: Agkistrodon bilineatus taylori,Agkistrodon bilineatus nisseolus andAgkistrodon bilineatus howardgloydi. These subspecies deviate in colour and distribution area from the nominate subspecies. The tropical moccasin is a sturdy venomous snake with a broad triangular head that is clearly distinct from the neck. On both sides of the head runs a bright white stripe which goes from the tip of the nose over the eye to the neck. Also from the tip of the nose ( on both sides) runs a second similar stripe parallel to the upper jaw, this terminates again in the neck region (hence bilineatus = with two stripes). The colour of the juveniles deviates from that of the adult animals. The pattern in juveniles consists of alternating light and dark brown bands ( several centimeters wide), each band is highlighted by a row of white scales. -

Rattlesnake Genome Supplemental Materials 1 SUPPLEMENTAL
Rattlesnake Genome Supplemental Materials 1 1 SUPPLEMENTAL MATERIALS 2 Table of Contents 3 1. Supplementary Methods …… 2 4 2. Supplemental Tables ……….. 23 5 3. Supplemental Figures ………. 37 Rattlesnake Genome Supplemental Materials 2 6 1. SUPPLEMENTARY METHODS 7 Prairie Rattlesnake Genome Sequencing and Assembly 8 A male Prairie Rattlesnake (Crotalus viridis viridis) collected from a wild population in Colorado was 9 used to generate the genome sequence. This specimen was collected and humanely euthanized according 10 to University of Northern Colorado Institutional Animal Care and Use Committee protocols 0901C-SM- 11 MLChick-12 and 1302D-SM-S-16. Colorado Parks and Wildlife scientific collecting license 12HP974 12 issued to S.P. Mackessy authorized collection of the animal. Genomic DNA was extracted using a 13 standard Phenol-Chloroform-Isoamyl alcohol extraction from liver tissue that was snap frozen in liquid 14 nitrogen. Multiple short-read sequencing libraries were prepared and sequenced on various platforms, 15 including 50bp single-end and 150bp paired-end reads on an Illumina GAII, 100bp paired-end reads on an 16 Illumina HiSeq, and 300bp paired-end reads on an Illumina MiSeq. Long insert libraries were also 17 constructed by and sequenced on the PacBio platform. Finally, we constructed two sets of mate-pair 18 libraries using an Illumina Nextera Mate Pair kit, with insert sizes of 3-5 kb and 6-8 kb, respectively. 19 These were sequenced on two Illumina HiSeq lanes with 150bp paired-end sequencing reads. Short and 20 long read data were used to assemble the previous genome assembly version CroVir2.0 (NCBI accession 21 SAMN07738522). -

A New Subspecies of the Pit Viper, Agkistrodon Bilineatus
PROC. BIOL. SOC. WASH. 97(1), 1984, pp. 135-141 A NEW SUBSPECIES OF THE PIT VIPER, AGKISTRODON BILINEAT US (REPTILIA: VIPERIDAE) FROM CENTRAL AMERICA Roger Conant Abstract. -A new subspecies ofthe cantil, Agkistrodon bilineatus howardgloydi, is reported from Costa Rica, H onduras, and Nicaragua. This snake is known locally as the castellana, and is distinguished from other subspecies of A. bilineatus by severa} pattem characteristics of the head and body and by a low number of ventral scales. During the preparation of a monograph (Gloyd and Conant) on the pit vipers, subfamily Crotalinae, of the genus Agkistrodon (sensu lato), I assembled virtually all the known rnuseum specimens of A. bilineatus from Central America. By comparison ofthese with numerous examples ofthe severa! subspecies indigenous to Middle America, the following facts emerged: (1) A. b. russeolus Gloyd occurs in disjunct subhumid areas of northern Belize, as suggested by Hoevers and Henderson (1974), H enderson and H oevers (J 975), and Henderson (1 978); (2) the range of the nominate subspecies, A. b. bilineatus Günther, extends south eastward from southern Sonora, Mexico, through the Pacific coasta l plain and versants of the adjacent mountains, and terminates in El Salvador; and (3) a distinct form, a specimen of which Cruz, et al. (1979), and Wilson and Meyer (1982) were unable to assign to any of the described taxa, occurs in Costa Rica, Honduras, and Nicaragua. This last forrn merits taxonomic recognition. Agkistrodon bilineatus howardgloydi, new subspecies Spanish name: Castellana Fig. 1 Holotype. -American Museum of Natural History (AMNH 125525), totallength 746 mm, male, collected by Louis W.